24. Quantification of peripheral benzodiazepine receptors in human brain with 18F-PBR06
Y. Fujimura, S. Zoghbi, F. Simeon, A. Taku, P. Victor, R. Innis and M. Fujita
Molecular Imaging Branch, National Institute of Mental Health, Bethesda, Maryland, USA
Objectives: The peripheral benzodiazepine receptor (PBR) is upregulated on activated microglia and macrophages and is, thus, a biomarker of inflammation. We previously reported that a 11C-labeled aryloxyanilide (T1/2 = 20 mins) was able to quantify PBRs in healthy human brain. Since many PET centers would benefit from a longer-lived 18F-labeled radioligand (T1/2 = 110 mins), the objective of this study was to evaluate the ability of a closely related aryloxyanilide (18F-N-fluoroacetyl-N-(2,5-dimethoxybenzyl)-2-phenoxyaniline; 18F-PBR06) to quantify PBRs in healthy human brain.
Methods: Nine human subjects were injected with about 185 MBq of 18F-PBR06 and scanned over five hours, with rest periods outside the camera. The concentrations of 18F-PBR06, separated from radiometabolites, were measured in arterial plasma.
Results: Modeling of regional brain and plasma data showed that a two-tissue compartment model was superior to a one-tissue compartment model, consistent with measurable amounts of both receptor-specific and nonspecific binding. Concentrations of brain activity measured with PET were consistently greater than the modeled values at late (280 to 300 mins) but not at early time points, which may have been caused by the slow accumulation of radiometabolite(s) in brain. To determine an adequate time for more accurate measurement of distribution volume (VT), which is the summation of receptor binding and nondisplaceable activity, we investigated which scan duration would be associated with maximal or near maximal identifiability. We found that a scan duration of 120 mins provided the best identifiability of VT: approximately two percent. The images showed no significant defluorination.
Time-activity data and curve fitting for temporal cortex. The two-compartment model provided significantly better fitting than the one-compartment model in all regions for all subjects.
Conclusion:18F-PBR06 can quantify PBRs in healthy human brain using 120 mins of image acquisition and concurrent measurements of radioligand in plasma. Although brain activity is likely contaminated with radiometabolites, the percentage contamination is thought to be small (<10%), since values of distribution volume are stable during 60 to 120 mins and vary by less than 10%. 18F-FBR is a longer-lived and promising alternative to 11C-labeled radioligands to measure PBRs as a biomarker of inflammation in the body.
Disclosure: This research was supported by the Intramural Program of NIMH (project # Z01-MH-002852-04). Y.F. was supported by the JSPS Research Fellowship in Biomedical and Behavioral Research at NIH.
29. Quantitative analysis of peripheral benzodiazepine receptor in human brain using PET with [11C]AC-5216
M. Miyoshi1,2, H. Ito1, R. Arakawa1, H. Takahashi1, H. Takano1, M. Okumura1, T. Otsuka1, F. Kodaka1, M. Sekine1, T. Sasaki1, S. Fujie1, C. Seki1, R. Nakao3, T. Fukumura3, K. Suzuki3, M. Matsumoto2 and T. Suhara1
1Molecular Neuroimaging Group, Molecular Imaging Center, National Institute of Radiological Sciences, Chiba; 2Department of Clinical Neuroscience and Therapeutics, Hiroshima University School of Medicine, Hiroshima; 3Molecular Prrobe Group, Molecular Imaging Center, National Institute of Radiological Sciences, Chiba, Japan
Objectives: Peripheral benzodiazepine receptor (PBR) is upregulated on activated glial cells and is therefore a useful biomarker of inflammation in the injured brain and neurodegenerative disorders, such as Alzheimer's disease.1,2 We developed a new PET radioligand, [11C]AC-5216, that allows the imaging and quantification of PBRs in monkey and mouse brains.3 [11C]AC-5216 can be applied to clinical investigation of PBR expression and therefore also microglia activation in neurological diseases. The aim of this study was to evaluate a quantification method of [11C]AC-5216 binding in human brain using PET measurements performed on healthy human subjects.
Methods: A 90-min dynamic PET scan was performed on each of twelve healthy men (age range 20 to 33 years; mean±s.d., 24.6±4.5) after intravenous injection of [11C]AC-5216. Regions-of-interest were drawn on several brain regions. Binding potential compared to non-displaceable uptake (BPND) was calculated by nonlinear least-squares fitting (NLS) method with the two-tissue compartment model, and total volume of distribution (VT) was also estimated by NLS and graphical analysis (GA) method. For these analyses, PMOD ver.2.8 was used.
Results: The distribution of radioactivity was widespread and fairly uniform in gray matter of the cerebral cortices and cerebellum, striatum, and thalamus. After intervenous injection of [11C]AC-5216, radioactivity peaked at about 2 to 3 mins, followed by slow washout. BPND was highest in the thalamus (4.6±1.0) and lowest in the striatum (3.5±0.7). VT values calculated by NLS were 6.9±1.8 and 4.9±1.5 mL/mL for the thalamus and striatum, respectively. K1/k2 values (K1: influx rate constant, k2: efflux rate constant) ranged from 1.3±0.4 to 1.6±0.3 mL/mL. The inter-individual variation of BPND was larger than VT, indicating that the inter-individual variation of VT is mainly caused by the variation of K1/k2 rather than BPND. VT obtained by NLS or GA showed similar regional distribution to BPND. However, there was no correlation between BPND and VT (VT by NLS, r = 0.31; VT by GA, r = 0.19) because of relatively larger inter-individual variation of K1/k2.
Conclusion: The regional distribution of [11C]AC-5216 was in good agreement with previous PET studies of PBRs in human brain, e.g., [11C]PK111954 and [11C]DAA1106.5 BPND is more appropriate for estimating [11C]AC-5216 binding than VT due to the inter-individual variation of K1/k2. [11C]AC-5216 is a promising PET ligand for quantifying PBR in human brain.
30. [18F]NS10743: a potential radiotracer for imaging of alpha-7 nicotinic acetylcholine receptors (α7-nAChRs)
P. Brust1, W. Deuther-Conrad1, S. Fischer1, A. Hiller1, P.-G. Hoffmeister1, C. Donat1, R. Bauer2, E. Østergaard Nielsen3, D. Brunicardi Timmermann3, O. Sabri4, J. Steinbach1 and D. Peters3
1Radiopharmacy, Institute of Interdisciplinary Isotope Research, Leipzig; 2Institute of Molecular Cell Biology, Friedrich Schiller University, Jena, Germany; 3NeuroSearch A/S, Ballerup, Denmark; 4Nuclear Medicine, University of Leipzig, Leipzig, Germany
Objectives: The α7-nAChR properties are impaired in schizophrenia, brain trauma and neurodegenerative diseases. Furthermore, α7-nAChRs are overexpressed in numerous tumor cells and represent a potential target for therapy. The outstanding diversity of cellular properties mediated by neuronal and non-neuronal α7-nAChRs points to the diagnostic potential of quantitative molecular imaging of α7-nAChRs. We have developed [18F]NS10743 (KD = 7.7 nmol/L), the first 18F-labeled radioligand for PET imaging of α7-nAChRs, and assessed the radiotracer selectivity in mice and pigs.
Methods: [18F]NS10743 was synthesised by nucleophilic substitution of the nitro precursor with specific activity >150 GBq/μmol and radiochemical purity >99%. Its biodistribution was determined in mice at 5, 20 and 60 mins p.i. To determine the specificity, separate animals were pre-treated with the α7 agonist SSR180711 (10 mg/kg). Ex vivo autoradiography was performed on mice brain at 60 mins after injection of 250 MBq [18F]NS10743. In vitro autoradiography was performed on porcine brain using [125I]α-bungarotoxin as α7 ligand. Furthermore, anesthetized piglets were intravenously injected with 250 to 500 MBq of [18F]NS10743. Selected animals additionally received 3 mg/kg SSR180711 at 10 mins prior to radiotracer application followed by a continuous infusion (2 mg/kg/2 h). Acquisition of dynamic PET scans (2 h) started with injection of [18F]NS10743. About 50 plasma samples were obtained, and plasma metabolites were analyzed with HPLC. Regions of interest (brain and eye) were drawn on summed PET images and distribution volumes were estimated.
Results: The brain uptake of [18F]NS10743 in mice was 4.8±0.4 %ID/g at 5 mins, 4.0±0.4 %ID/g at 20 mins, and 1.6±0.3 %ID/g at 60 mins p.i. High initial radioactivity uptake was also observed in adrenal glands (11.3 %ID/g), spleen (7.7 %ID/g), pancreas (6.0 %ID/g), and thymus (4.4 %ID/g). Organ distribution of mice pre-treated with SSR180711 shows a significant reduction of radioactivity uptake at 60 mins p.i. in adrenals, brain, thymus, and pancreas, all organs which express α7-nAChRs. Ex vivo autoradiography revealed a pattern of [18F]NS10743 binding in anatomically defined brain structures such as hippocampus and cortex which closely matches regions with high α7-nAChRs expression. Summed PET images obtained in piglets show a distribution pattern similar to in vitro autoradiographs of [125I]α-bungarotoxin with high radiotracer binding in cortex, moderate binding in cerebellum and low binding in white matter. The brain uptake is comparable to that of the α4β2 nAChR-ligand 2-[18F]F-A-85380. Application of SSR180711 resulted in more than two-fold increase of brain uptake. However, the distribution volume was decreased by about 20%, indicating inhibition of specific radiotracer binding. Furthermore, the flow-independent uptake of [18F]NS10743 in eye, which is known to express α7-nAChRs, was reduced by about 30%. Metabolite studies revealed the presence of a single major metabolite in mice and piglets, which is probably the result of an enzymatic oxidation of the nitrogen at position 1 in the diaza-bicyclononane moiety of NS10743.
Conclusions: [18F]NS10743 is a high affinity ligand for α7-nAChRs with good brain uptake, specific receptor binding in vivo and good metabolic stability, which makes it suitable for neuroimaging with PET.
124. Biodistribution and radiation dosimetry of the NK1 ligand GR-205171 determined from Papio Anubis whole-body PET
K. Ridler1, E.A. Rabiner1, L. Kegeles2, J. Castrillon2, E. Hackett2, M. Laruelle1 and M. Slifstein2
1Clinical Imaging Centre, Glaxosmithkline, London, UK; 2Division of Translational Imaging, Department of Psychiatry, Columbia University, New York, New York, USA
Background and aims: [11C]GR-205171 ([2(11C)-methoxy-5-(5-trifluoromethyl-tetrazol-1-yl)-benzyl]-(2S-phenyl-piperidin-3S-yl)-amine) is a selective neurokinin 1 (NK1) receptor PET ligand. Positron emission tomography (PET) was used to determine the whole body radiation dosimetry of [11C]GR-205171, in 3 adult male baboons (Papio Anubis).
Methods: Three anaesthetized male baboons (Papio Anubis), weight 25.4±2.3 kg) were examined on a Siemens Accel whole body scanner in 2D mode for 85 mins following a bolus injection of [11C]GR-205171 (185±26 MBq). Subjects were scanned at 6 overlapping bed positions covering from head to mid-thigh. Organs with activity above background level were identified on the reconstructed PET images. Time activity curves were generated from sub-samples of these organs and area under the curve was estimated (Bq/cm3*hr). These values were multiplied by organ volumes of a 70 kg mathematical phantom,1 normalized for body mass and injected dose to obtain residence times (hr). Residence times were used as input to the Olinda program2 to generate estimates of absorbed dose and Equivalent Dose (ED).
Results: Individual organ absorbed doses are presented in Table 1 (mSv/MBq). Effective dose was estimated at 8.72±2.23 μSv/MBq. The dose limiting organ was the lung (5.54 × 10−2 mSv/MBq). For studies performed under RDRC, this would limit single study dose to a maximum of 888 MBq.
Conclusions: We estimated [11C]GR-205171 radiation dosimetry from data acquired with whole-body PET in 3 non-human primates. The estimated effective dose was 8.72±2.23 μSv/MBq, within the range of carbon-11 labelled ligands. A 370MBq injection of [11C]GR-205171 would result in an exposure of 3.2 mSv (excluding exposure from accompanying transmission scanning).
High levels of absorbed radiation in the lung may be due to specific binding of [11C]GSK-205171. Examination following a high dose of an NK1 blocking compound, will be important, in assessing the effects of peripheral and central NK1 blockade on estimated dosimetry figures.
Radiation-Absorbed Dose Estimates (mSv/MBq)
Organ
Mean (s.d.)
Organ
Mean (s.d.)
Organ
Mean (s.d.)
Adrenals
3.46E−03 (3.21E−05)
Kidney
1.48E−02 (1.60E−03)
Spleen
2.31E−02 (3.15E−03)
Brain
5.64E−03 (1.21E−03)
Liver
1.10E−02 (6.11E−04)
Testes
9.89E−04 (2.67E−05)
Breasts
2.42E−03 (1.71E−04)
Lungs
5.54E−02 (2.15E−02)
Thyroid
2.86E−03 (4.69E−04)
Gallbladder Wall
2.76E−03 (4.74E−04)
Muscle
1.53E−03 (2.95E−04)
Urinary bladder wall
1.40E−03 (5.09E−04)
Lower large intestine wall
1.01E−03 (6.03E−04)
Ovaries
1.15E−03 (6.15E−04)
Uterus
1.12E−03 (6.18E−04)
Small intestine
3.69E−03 (2.41E−03)
Pancreas
3.25E−03 (1.80E−04)
Total body
2.79E−03 (2.08E−05)
Stomach wall
2.21E−03 (2.92E−04)
Red marrow
1.79E−03 (1.68E−04)
Upper large inetestine wall
1.60E−03 (5.98E−04)
Osteogenic Cells
1.85E−03 (5.76E−04)
Effective dose equivalent
1.21E−02 (2.69E−03)
Heart wall
1.86E−02 (4.47E−03)
Skin
1.01E−03 (3.29E−04)
Effective dose
8.72E−03 (2.32EE−03)
262. Parametric mapping of NK1 receptor binding using [11C]R116301
S. Wolfensberger, M. Yaqub, B. Van Berckel, B. Windhorst, J. Leysen, A. Lammertsma and R. Boellaard
Nuclear Medicine & PET Research, VU University Medical Center, Amsterdam, The Netherlands
Purpose: NK1 receptors have been implicated in various neuropsychiatric disorders. [11C]R116301 has been proposed as a PET tracer for visualizing and quantifying NK1 receptors in vivo. In previous studies, both simplified reference tissue models and tissue ratio methods have been used, in combination with a region of interest (ROI) approach. The purpose of this study was to determine the optimal method for parametric analysis of [11C]R116301 binding.
Methods: Two dynamic PET studies were performed in 11 normal volunteers. Three subjects were scanned before and after a blocking dose of aprepitant and 8 subjects were scanned twice under baseline conditions (test-retest). Total scan duration was 90 mins and for all subjects scans were separated by 5 h. Data were analysed using striatum as region of interest and cerebellum as reference tissue. Parametric binding potential (BPND) images were obtained using receptor parametric mapping (RPM), a basis function implementation of the simplified reference tissue model (SRTM), five versions of a multi-linear reference tissue method (MRTM), and Logan graphical analyses with reference tissue input. In addition, striatum to cerebellum standardized uptake value ratio minus 1 (SUVr-1) for the time interval 60 to 90 mins, an approximation for BPND, was evaluated. Results of all these methods were compared with BPND derived using SRTM. Finally, effects of shortening scan duration on BPND were evaluated.
Results: When comparing kinetic methods with SRTM, best performance was obtained with RPM (fitting for R1, k2 and BPND). This method showed high correlation (r2 = 0.93 and 0.82, respectively), with acceptable bias (regression slopes 1.10 and 0.88, respectively). RPM also provided good test-retest variability (VAR = 10.1±8.9% and 12.6±5.5%, respectively) with good reliability (ICC = 0.78 and 0.86, respectively). Linearized methods, i.e. all MRTM versions and reference Logan, failed to provide accurate and precise estimates of BPND, probably due to the slow kinetics of this tracer. Surprisingly, SUVr-1 also showed high correlation with SRTM (r2 = 0.96, regression slope = 0.85) with both excellent test-retest VAR (6.2%±3.07) and ICC (0.91). After reducing analysis time to 60 mins, RPM correlation with 90 mins SRTM remained high (r2 = 0.87, regression slope = 0.90). There was, however, some decline in test-retest VAR (17.13%±14.79) and reliability (ICC = 0.62). In addition, there was only a small reduction in performance of SUVr-1 when the time interval was reduced to 80 to 90 mins.
Conclusion: RPM is the optimal parametric method for quantifying [11C]R116301 binding. It shows both good test-retest variability and reliability, and there is scope for reducing scan times. A simple tissue ratio method also holds promise for routine clinical applications.
279. Imaging and quantitation of cannabinoid CB1 receptors in healthy human brain using the inverse agonist radioligand [11C]MePPEP
G. Terry1,2, J.-S. Liow1, S. Zoghbi1, J. Hirvonen1, A. Farris1, A. Lerner1, J. Tauscher3, J. Schaus3, L. Phebus3, C. Felder3, C. Morse1, J. Hong1, C. Halldin2, V. Pike1 and R. Innis1
1Molecular Imaging Branch, National Institute of Mental Health, Bethesda, Maryland, USA; 2Department of Clinical Neuroscience, Psychiatry Section, Karolinska Institutet, Stockholm, Sweden; 3Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, Indiana, USA
Objectives: [11C]MePPEP ((3R,5R)-5-(3-methoxy-phenyl)-3-((R)-1-phenyl-ethylamino)-1-(4-trifluoromethyl-phenyl)-pyrrolidin-2-one) is a positron emission tomography (PET) radioligand for imaging and quantifying cannabinoid CB1 receptors that has been studied in rodents1 and monkeys.2 The goals of this study were to compare the simple outcome measure of brain uptake (determined from 40 to 80 mins) with distribution volume (determined from brain data and serial concentrations of parent radioligand separated from radiometabolites in plasma) in healthy human brain. Although our initial scans lasted for 150 mins, we later extended the acquisitions to 210 mins to ensure that time-independent measures of distribution volume were stable and well identified.
Methods: [11C]MePPEP was studied in 16 subjects with 23 PET scans (643±95 MBq) using a GE Advance camera (GE Healthcare). We examined the ability of [11C]MePPEP to image and quantify CB1 receptors by using the ‘gold standard’ of serial measurements of unchanged parent in arterial plasma for up to 120 mins and brain radioactivity for up to either 150 mins (n = 8) or 210 mins (n = 8) after injection. We examined the retest reliability (i.e., within subject variability) of our measurements by conducting test/retest studies (n = 7), as well as the intersubject variability (i.e., coefficient of variability = s.d./mean) of our measurements.
Results: The brain uptake of [11C]MePPEP was high (200 to 400% SUV) and relatively stable, which allowed images to be acquired for 210 mins (Figure 1). A two-tissue compartment model provided a better fit of the measured data than a one-tissue compartment model. Distribution volume (VT) was well identified (5%) and provided stable values from 60 mins until the end of 210 mins of scanning. We observed a moderate intra- and intersubject variability of brain uptake (7% and 18%), and a higher intra- and intersubject variability of VT (13% and 55%).
Time-activity curves of [11C]MePPEP in brain. Measurements from the putamen (▪), prefrontal cortex (□), cerebellum (•), pons (○), and white matter (X) were fitted with an unconstrained 2-tissue compartment model (—). Data from this subject is representative of all subjects. Conc = concentration.
Conclusions: The distribution volume (VT) of [11C]MePPEP was well measured by the standard criteria of identifiability, time stability, and moderate retest variability. However, the intersubject variability of VT for [11C]MePPEP was much higher than that of the simple measure of brain uptake between 40 and 80 mins in this small sample size. We do not know the true variability of CB1 receptor density in this sample. Nevertheless, in comparison to brain uptake, we suspect that VT is more accurate (since it corrects for exposure of the radioligand to the brain), but is less precise (because of the noise added by the plasma measurements and modeling analysis). Thus, both brain uptake and VT should be used with caution in any clinical studies, since neither has both high accuracy and high precision.
455. Protoporphyrin IX interaction with peripheral type benzodiazepine receptor in rats
H. Ozaki, S. Zoghbi, J. Hong, V. Pike, R. Innis and M. Fujita
Molecular Imaging Branch, National Institute of Mental Health, Bethesda, Maryland, USA
Objectives: The major physiological porphyrin, protoporphyrin IX (PPIX), is well-known to bind to peripheral type benzodiazepine receptor (PBR) as one of the endogenous ligands.1 The published Ki value of PPIX ranges from 0.015 to 19 μmol/L, whereas the other porphyrins are less potent at PBR.1,2 The purpose of this study was to evaluate PPIX interaction with PBR in vivo using PET. We used 5-aminolevulinic acid (ALA) for this study because with its administration to rats, plasma concentration of PPIX is expected to become in the range of its Ki value for PBR.3
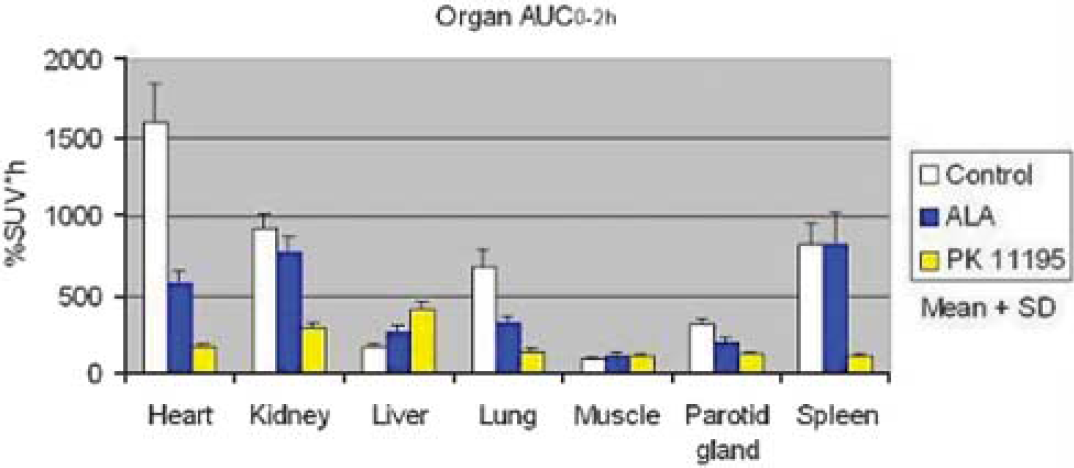
Methods: Male Sprague-Dawley rats (about 320 g body weight) were divided into 3 groups (control, ALA- and PK 11195-pretreated groups: n = 4, 4 and 3, respectively). ALA (200 mg/kg) was intravenously injected to animals 2 h before PET scans. This dose of ALA produces plasma and organ PPIX peak levels of 0.5 to 20 μmol/L.3 For comparison, a receptor saturating dose of PK 11195 (10 mg/kg) was intravenously administered 2 mins before PET scans. In each animal, one 2 h PET scan of PBR was performed using N-acetyl-N-(2-[11C]methoxybenzyl)-2-phenoxy-5-pyridinamine ([11C]PBR28) and the HRRT device under isoflurane anesthesia. The time activity curves of the heart, kidney, liver, lung, skeletal muscle, parotid gland and spleen were drawn using individual regions of interest and then the area under the curve for each organ was calculated. In 2 animals of each of the 3 groups, more quantitative analysis was performed in brain by measuring total distribution volume using an unconstrained two-tissue compartment models and metabolite-corrected arterial input function.
Results: Control scans with [11C]PBR28 showed high levels of uptake in the heart, kidney, lung and spleen. The ALA-pretreatment dramatically suppressed the normal uptake of radioactivities in the heart, lung and parotid gland (Figure 1, P<0.01). However, there was no blocking effect in the kidney, liver, skeletal muscle and spleen (Figure 1). PK 11195 also blocked the radiotracer binding to PBR in the heart, kidney, lung, parotid gland and spleen (Figure 1, P<0.01). In the brain, the distribution volume was decreased by both pretreatments. In all organs, blocking effect of 200 mg/kg of ALA was weaker than that of 10 mg/kg of PK 11195.
Area under curve of each organ for 2 h after [11C]PBR28 injection in the control. ALA-pretreated and PK 11195-pretreated groups.
Conclusions: PPIX reduced [11C]PBR28 binding to PBR in rats although 200 mg/kg of PPIX was not enough for complete blocking. This study provides the first indication for the in vivo competitive binding of PPIX with a PET ligand at PBR. Since endogenous PPIX levels are high in porphyria, this disorder should demonstrate reduced target uptakes of the radiotracer as seen in the rats.
457. [11C]-CIMBI5: a novel 5-HT2A agonist PET tracer
A. Ettrup1, M. Palner1, N. Gillings2, K. Någren2, L.K. Rasmussen3, S. Keller2, M. Sibomana2, M. Begtrup3, J. Madsen2 and G.M. Knudsen1
1Neurobiology Research Unit and Center for Integrated Molecular Brain Imaging (CIMBI); 2PET and Cyclotron Unit, Copenhagen University Hospital, Rigshospitalet; 3Department of Medicinal Chemistry, Faculty of Pharmaceutical Sciences, University of Copenhagen, Copenhagen, Denmark
Objectives: Receptor agonist PET tracers have a better potential than antagonist tracers to reflect displacement under endogenous neurotransmitter release, however, only antagonistic PET tracers targeting the 5-HT2A receptor are currently known. The aim of this study was to validate a novel 5-HT2A agonist PET tracer in the pig brain.
Methods: The high-affinity 5-HT2A receptor selective agonist N-(2-[11C-OCH3]methoxybenzyl)-2,5-dimethoxy-4-iodophenethylamine ([11C]-INBMeO, [11C]-CIMBI5) was radiolabelled by methylation of the N-Boc-protected precursor using [11C]methyl triflate and subsequent deprotection with TFA. In five Danish Landrace pigs, [11C]-CIMBI5 was given as IV bolus injection, and the pigs were subsequently PET scanned with a HRRT camera. Three of the pigs were scanned a second time, now under treatment with the 5-HT2A receptor antagonist ketanserin (3 mg/kg bolus, 1 mg/kg*hour infusion). Total activity in full blood, plasma, and radioactive metabolites were measured throughout the scan using HPLC.
Results: Compared to cerebellum [11C]-CIMBI5 showed a high cortical uptake, and the time activity curves confirmed significantly higher uptake in cortex compared to cerebellum showing a ratio (AUCcortex/AUCcerebellum) of approximately 1.4. Following ketanserin pre-treatment, the cortical binding of [11C]-CIMBI5 was reduced to cerebellar levels. In the radio-HPLC analysis, we found a lipophilic radioactive metabolite building accounting for up to 20% of the total activity in plasma and maintaining stable levels after 20 mins and throughout the scan.
[11C]-CIMBI5 time acitivity curves.
Conclusions: The novel agonist PET tracer [11C]-CIMBI5 distributes in the brain in a pattern compatible with the known 5-HT2A receptor distribution, and its binding is displaceable by ketanserin. Thus, [11C]-CIMBI5 is a promising candidate for human 5-HT2A PET scanning, although the relatively high cerebellum binding, indicative of a high non-specific binding, may compromise the possibility to detect changes in [11C]-CIMBI5 binding following changes in extracellular 5-HT levels. Therefore, changing the 11C-labeling site to eliminate the lipophilic metabolite or changing the chemical structure to alter lipophilicity may optimize the signal-to-noise ratio and thus improving the potential of [11C]-CIMBI5 for future clinical use.
466. Development of dPET, a PET technique to measure the diffusion of drugs after direct delivery to the brain
R.W. Sirianni1, W.M. Saltzman2 and R.E. Carson1,2
1Diagnostic Radiology; 2Biomedical Engineering, Yale University, New Haven, Connecticut, USA
Objectives: Active agents have been delivered directly to the brain to treat or better understand diseases in the fields of substance abuse, mental illness, neurodegenerative disorders, brain cancer, ischemia and epilepsy, yet the spatial gradients that result from local delivery are not always known. Methods have been developed to measure drug transport parameters in vitro through use of brain slices or extracted tissue elements, however, these methods do not capture the full complexity of transport in living tissue, where the diffusion of drugs occurs in parallel with binding and uptake by cells, elimination and degradation. Methods used to measure drug distribution in vivo are too invasive to apply to humans and therefore cannot monitor the effectiveness of drug delivery in clinical studies. In these studies, we developed a method for non-invasive measurement of the diffusion of radiotracers with Positron Emission Tomography (PET). This diffusion-PET (dPET) system will eventually be used to characterize the spatial distribution of radiolabeled drugs or drug-polymer delivery systems after direct delivery in vivo.
Methods: The rate of movement of bound (Cb)and free drug (Cf) from a point source may be expressed in two partial differential equations that are a function of radial position (r), time (t), the diffusion coefficient (D), and a source term that accounts for non-diffusional processes such as binding, uptake or elimination. A numerical solution for the total detectable concentration of drug can be generated by assuming uniform distribution of first-order elimination (k2) and reversible binding to cells (kon, koff). dPET data were simulated in Matlab by accounting for (1) decay, (2) counting statistics, and (3) resolution (1.5 mm). Multiple data fitting trials were performed via least-squares to obtain an estimate for D in the presence of varying levels of binding and elimination. For initial in vitro tests, a point source of [18F]FDG(5 to 50 μl, <200 μCi) was introduced into an agarose tissue phantom. Data were collected on the microPET Focus 220 for 1 to 6 h, reconstructed via OSEM methods, and image data were analyzed to obtain an estimate of the diffusion coefficient.
Results: In modeling simulations, diffusion coefficients were accurately estimated for low molecular weight drugs subject to diffusion (D = 0.06 mm2/min), elimination (k2 = 0.1 min−1) and reversible binding (kon = 0.3 min−1 and koff = 0.075 min−1). Assuming 20% pixel-noise, accurate estimates of Deff were produced for both [18F] and [11C] in a 30 mins simulation (Dfitted = 0.060±0.0024 and 0.057±0.0030, respectively, n = 10).
Initial measurements of the [18F]FDG diffusion in an agarose brain phantom resulted in estimates of D (0.034 and 0.071 mm2/min) near its theoretical value (0.052 mm2/min). We expect that optimization of the infusion technique (e.g., preventing backflow) will improve parameter estimates.
Conclusions: Data-fitting experiments and preliminary in vitro measurements demonstrated the feasibility of using PET to measure the distribution of radiolabeled drugs after direct delivery to the brain. The studies described here provide the basis for the next step in the development of dPET, i.e., the measurement of tracer diffusion in vivo, and suggest that dPET is a viable method to measure the spatial distribution of radiolabeled drugs in the brain.
Sample in vitro data.
487. Kinetic modelling of a slow radioligand: [11C]GR205171 binding to NK1 receptors before and after administration of Casopitant
V.J. Cunningham1, E.A. Rabiner1, S.A. Fernandes2, R.N. Gunn1,3, R. Gomeni4 and S. Zamuner4
1GlaxoSmithKline Clinical Imaging Centre, Imperial College, London, UK; 2Discovery Medicine, Neurosciences Centre for Excellence In Drug Discovery, GlaxoSmithKline, Verona, Italy; 3Department of Engineering Science, Oxford University, Oxford, UK; 4Clinical Pharmacology, Modeling & Simulation, GlaxoSmithKline, Verona, Italy
Objectives: The PET radioligand, [11C] GR205171, is a high affinity, selective NK1-receptor antagonist that has been used in vivo to characterise NK1-receptor binding in rhesus monkeys1,2 and in humans.3 The kinetic behaviour of this radiotracer varies significantly across brain regions, consistent with the known distribution of NK1-receptors, but, because of slow kinetics, care is required in the selection of a PET model suitable for estimation of the correspondingly wide range of regional binding potentials.3 Casopitant is a selective NK1 antagonist currently in development for prevention of chemotherapy-induced and postoperative nausea and vomiting. As part of a study in humans to develop a pharmacokinetic-receptor occupancy (PK-RO) model for Casopitant, the suitability of several compartmental models for the estimation of [11C]GR205171 NK1 receptor binding was investigated in baseline scans and following administration of Casopitant.
Methods: Eight healthy male volunteers entered and completed a study to estimate the brain NK-1 RO following Casopitant. All subjects had 2 PET scans, at baseline and 24 h following a single dose of 2.5, 5, 20, 50, or 120 mg Casopitant. [11C]GR205171 was administered as an intravenous bolus. Arterial blood was collected throughout the scans and plasma samples were assayed for radiolabelled metabolites, enabling generation of arterial plasma parent input functions (IF). Eight regions of interest (ROI) were defined in each subject. Several modelling approaches were compared. One and two-tissue compartmental models, with reversible and irreversible kinetics and IF, and a reference tissue model with cerebellum as input were applied to each ROI in each individual scan independently. Finally analyses using compartmental models with simultaneous estimation of multiple regions sharing common parameters across regions and/or across regions and across scans within subjects, were applied.
Results: Analyses confirmed cerebellum as a reference region, albeit with two apparent tissue compartments. Estimates of RO at higher doses of Casopitant were consistently high (>85%) for all models. However there was variation between models in RO estimates at lower doses, reflecting different biases and stability. Models were compared on the basis of goodness of fit criteria, consistency of parameter estimates between regions and subjects, and on the pharmacological reasonableness of observed differences in the estimated rate constants. The most robust and consistent estimates were obtained using shared parameter models.
Conclusions: [11C]GR205171 has slow kinetics, in the context of an 11C PET scan, which presents difficulties in the accurate estimation of the receptor dissociation rate constant (k4), and hence leads to significant variability in the estimation of low RO. A shared parameter model which assumes that kinetic differences between regions and scans is attributable only to differences in plasma clearance (K1) and the association rate constant (k3), but with k4 and other parameters being the same across regions and scans, enhances the stability of the RO estimate and was considered most suitable for the analysis of [11C]GR205171 human PET studies.
536. In vitro quantification of mGluR5 in pons and cerebellum of human brain using [3H]ABP688
L. Minuzzi1, M. Diksic2, S. Gauthier3, R. Quirion4 and P. Rosa-Neto1
1Translational Neuroimaging Laboratory—MCSA, Douglas Hospital; 2Department of Neurology and Neurosurgery; 3McGill Centre for Studies in Aging, Douglas Hospital; 4Department of Psychiatry, Douglas Hospital, McGill University, Montreal, QC, Canada
Objectives: Metabotropic glutamate receptor type 5 (mGluR5) has been associated to the memory processing and also to several neuropsychiatric disorders. The radiopharmaceutical [11C]ABP688 has been showed to be a suitable PET ligand for measuring in vivo mGluR5 in animals and humans. However, despite the favourable kinetic, arterial blood sampling is required, implicating in technical limitations for PET imaging. The objective of the present study is to quantify the total number of mGluR5 in two regions of human brain that are potentially devoid or with markedly low receptors in order to provide a reference region for PET analysis. In vitro quantitative autoradiography was carried out using [3H]ABP688 in human cryosections of pons and cerebellum in comparison to a mGluR5-rich region hippocampus.
Methods: Frozen human brain section of hippocampus, pons and cerebellum from five healthy controls were cryosectioned at 20 μm. Saturation binding study was carried out according to the method described by Hintermann et al.2 with some modifications. Briefly, the tissue was pre-incubated for 20 mins in buffer containing 30 mmol/L Na HEPES, 110 nmol/L NaCl, 5 mmol/L KCl, 2.5 mmol/L CaCl2 and 1.2 mmol/L MgCl2 (pH 7.4). The brain sections were then incubated for one hour in the presence of [3H]ABP688 (74 Ci/mmol) at a range of concentrations from 0.125 to 8 nmol/L. Non-specific binding was determined with addition of 10 μmol/L MPEP. The slides were washed (3 × 5 mins) in cold buffer, dipped in ice-cold distilled water and dried. The sections along with autoradiographic standards were exposed for five days to phosphor imaging plates. The Bmax and KD of [3H]ABP688 were calculated by saturation binding analysis.
Results: Binding of [3H]ABP688 showed a heterogeneous and displaceable distribution in human tissue. Hippocampus (CA1) presented higher density of receptors (Bmax 1.31±0.07 pmol/mg of tissue). Sections of pons and cerebellum showed markedly lower Bmax (0.08±0.01 and 0.09±0.01 pmol/mg, respectively) in comparison to hippocampus (ratio 1:15). Dissociation constant (KD) of pons and cerebellum were identical 0.4±0.2 nmol/L, whereas hippocampus revealed KD of 1.5±0.2 nmol/L.
Conclusions: Saturation binding parameters in human tissue were similar to values found previously in rat brain using the same radioligand. Although pons and cerebellum showed presence of mGluR5, the distinctly lower amount of receptors suggests that they can be used as reference region for in vivo quantification of mGluR5 in humans.
Saturation Binding of [3H]ABP688 in Human Brain.
659. C-fibre LTP is associated with dynamic changes in opioid neurotransmission in the brain
T. Hjornevik1, B. Schoultz2, J. Gjerstad3,4, G. Henriksen2,5 and F. Willoch1,6
1Department of Anatomy & CMBN, University of Oslo; 2Department of Chemistry, University of Oslo; 3National Institute of Occupational Health; 4Department of Molecular Bioscience, University of Oslo, Oslo, Norway; 5Nuklearmedizinische Klinik and Poliklinik, Klinikium rechts der Isar, Technical University Munich, Munich, Germany; 6Department of Radiology, Aker University Hospital, Oslo, Norway
Objectives: Neuronal events leading to development of long-term potentiation (LTP) in the nociceptive pathways may be a cellular mechanism underlying hyperalgesia. Opioid receptors control the descending modulatory system to the spinal cord, and play an important role in anti-nociception. In the present study, we examine how induction of spinal LTP affects the supraspinal opioidergic system.
Methods: All animals were for the following experiments anesthetized with isoflurane gas. The left sciatic nerve was given a high-frequency conditioning stimulation (HFS) to induce LTP (5 trains/1 secs, 100 Hz, 1 ms pulses, 10 secs intervals):
Spinal field potential recordings. Field potentials were recorded from neurons at depths of 100 to 300 μm from the surface of the spinal cord (n = 6).
PET measurements.
PET studies were performed in parallel. Data was acquired (Inveon, Siemens) for 60 mins after i.v. injection of [11C]PEO, a u- and k-OR tracer. All rats (n = 8) underwent the first day a rest study without any intervention. The other day tracer acquisition was performed 2.5 h after HFS conditioning. The data was 3DRP reconstructed and binding potentials (BPND) were calculated using a reference tissue model (MRTM0) in PMOD (PMOD Technologies Ltd., Zurich, Switzerland). Statistical comparisons between baseline and stimulation condition were performed with SPM5 (Wellcome Department of CognitiveNeurology, Institute of Neurology, London, UK) after anatomic standardization to an average PET uptake template. Level of significance was set at P<0.05. The result image (HFS>baseline) was registered to a 2-D digital version of the Rat Brain atlas by Paxinos and Watson,1 and statistical values were extracted from predefined volumes of interest (VOIs).
Results: An increase in C-fibre response and reduced C-fibre threshold were observed following sciatic nerve HFS throughout the observation time of 3 h. Increased PEO binding was observed ipsilaterally in the amygdala, hippocampus, somatosensory cortex, and the superior colliculus. In addition, significant increased signal was located bilaterally in the nucleus accumbens, caudate putamen, and the hypothalamus.
Conclusions: The data shows that HFS applied to the sciatic nerve lead to a long lasting C-fibre LTP representative for hyperalgesia. Concomitantly, HFS was associated with a regional increased opioid receptor availability that is interpreted as reduced opioid tonic activity. The involved structures, amygdala in particular, are part of a pain modulatory circuitry. Therefore, a reduced descending opioid, anti-nociceptive activity may be related to the observed C-fibre LTP and associated abnormal pain states, such as hyperalgesia. Furthermore, the dynamic changes in opioid neurotransmission in the limbic structures may connect the findings with an affective-emotional response to the applied noxious stimulus.
671. Effects of a chronic exposure to nicotine: a study in baboon by using a multi-injection PET study with [18F]fluoro-A-85380
M. Bottlaender, N. Miro-Bernie, M.-A. Peyronneau, F. Dolle, S. Bourgeois, F. Hinnen, S. Goutal, W. Saba, J. Delforge and H. Valette
CEA, DSV, I2BM, Service Hospitalier Frederic Joliot, Orsay, France
Introduction: A particular feature of nicotinic acetylcholine receptors (nAChR) is that chronic exposure to nicotine induces a higher level of nicotine binding, termed up-regulation. The mechanism through which nicotine induces nAChR up-regulation is complex and not fully clarified to date. This feature has been evidenced in cells expressing nAChRs, rodents, monkeys and in humans. In the development of our program to study tobacco addiction, we aimed at studying in vivo this up-regulation. For this purpose, one Papio anubis baboon received nicotine continuously (Alzet osmotic minipumps) for 6 months. At various times (15 days, 2, 3, 4 and 6 months), the minipumps were removed and the baboon underwent, two or three days later, a multi-injection PET experiment.
Methods: The compartment model describing the fluoro-A-85380 kinetics is the usual non-equilibrium non-linear model. It includes four compartments and seven parameters. The PET protocol included three injections: a tracer injection of [18F]fluoro-A-85380 (time 0), a partial saturation injection (90 mins) obtained by a simultaneous injection of labelled and unlabelled fluoro-A-85380 (35 nmol), and a saturation injection (180 mins) by a large amount of only unlabeled fluoro-A-85380 (1500 nmol). Measurements of plasma nicotine concentration during PET experiments allowed performing a correction, if needed (concentration >1 ng/mL), by introducing in the model the nicotine kinetics. The nicotine parameters were set to values estimated in a previous study (Bottlaender, NeuroImage 2008). This method allowed to simulate the effect of residual nicotine concentration in the brain, to correct the ligand kinetic and therefore to estimate the ligand parameters without significant bias.
Results: During nicotine administration, the mean plasma nicotine concentration was 10 to 20 ng/mL. The estimation of nicotinic receptor concentration in thalamus remained unchanged: 4.66±0.58 pmol/mL from 15th day to 6th month versus control: 4.87 pmol/mL, and 4.44 pmol/mL after three weeks of weaning. In contrast, the estimated KdVr, was twice smaller, 0.24±0.07 nmol/L, than the control value: 0.61 nmol/L. After three weeks of weaning, KdVr returned to control value (0.63 nmol/L). The estimates of the other model parameters were not disturbed by the chronic nicotine administration. However, this present approach can be used mainly in receptor-rich regions because difficulties in estimating all model parameters in receptor-poor regions.
This observed variation in nicotinic receptor affinity is in agreement with the usual response to the administration of an agonist, which induces conformational changes from low to high affinity state of the nAChR (Edelstein, Biol Cybern 1996). In contrary to published data, an up-regulation of nAChR in thalamus is not observed. However, our results are in agreement with one in vitro study (Vallejo J Neurosci. 2005).
Conclusions: Other previous studies in smokers had shown an increase of distribution volume and/or binding potential (Staley, J Neurosci 2006; Mukhin;. J Nucl Med 2008). Our results are in agreement with these findings, but show that this increase is only a consequence of an affinity change, all the other parameters, including the nicotinic receptor concentration, remaining unchanged.
Acknowledgments: This research was supported by grant from ‘Canceropole, Ile-de-France, projet PL026’.
683. Kinetic parameters of BAY 94–9172 binding to β-amyloid in human brains computed from PET data
G. Becker1, H. Barthel1, M. Patt1, J. Luthardt1, A. Seese1, E. Hammerstein2, B. Eggers3, C. Reininger4, B. Rohde4, U. Hegerl2, H.-J. Gertz2 and O. Sabri1
1Department of Nuclear Medicine; 2Department of Psychiatry, University Hospital Leipzig; 3Arzneimittelforschung Ltd., Leipzig; 4Bayer-Schering Pharma AG, Berlin, Germany
Objectives: BAY 94–9172 is an 18F-labeled stilbene derivative with a high affinity and specificity for β-amyloid in vitro. In early phase clinical development the visual assessment of BAY 94–9172 PET brain scans has demonstrated convincing diagnostic accuracy in the differentiation between subjects with Alzheimer's disease (AD) and healthy volunteers (HV) [Rowe et al, Lancet Neurol 2008, Barthel et al., J Nucl Med 2008]. Modeling of the kinetics of this PET tracer provides parameters by which direct quantification of brain β-amyloid load can be acheived. The potential for these parameters for distinguishing between AD subjects and HVs was investigated.
Methods: After intravenous administration of 300 MBq BAY 94–9172, the PET brain imaging was performed in 7 AD patients and 10 age-matched HVs using an ECAT EXACT HR+ system in 3D-acquisition mode. 23 frames were acquired from 0 to 90 mins post injection. In each subject imaged, kinetic modeling was applied to the volume of interest (VOI) based tissue-activity curves generated for 25 brain regions (anatomically defined via MRI co-registration) using a per subject metabolite-corrected arterial input-function. Total distribution volume (DV) and binding potential (BP) = k3/k4 and k3 were used to characterize specific binding. Both standardized uptake value ratio (SUVR) and distribution volume ratio (DVR) were computed using the cerebellar cortex as a reference region.
Results: All cortical regions and the cerebellar cortex need two tissue compartments to be described adequately. All investigated parameters were significantly higher in ADs compared to HVs e.g. in frontal cortex (DV: 11.3±3.5 versus 7.2±1.3, P = 0.004; BP: 3.2±1.3 versus 1.6±0.3, P = 0.002; k3 = 0.059±0.029 versus 0.028±0.005, P = 0.004), parietal cortex (DV: 10.2±2.3 versus 7.2±1.2, P = 0.004; BP: 3.0±1.0 versus 1.6±0.2, P = 0.0004; k3 = 0.049±0.022 versus 0.028±0.006, P = 0.01) and posterior cingulate cortex (DV: 11.7±3.1 versus 7.5±1.2, P = 0.002; BP: 3.4±1.3 versus 1.7±0.2, P = 0.001; k3 = 0.062±0.029 versus 0.027±0.007, P = 0.002). When the cerebellar cortex was used as a reference region (DVR, SUVR) results were highly significant (P<0.0001) for all three regions, too.
Conclusions: Kinetic modeling of BAY 94–9172 brain PET images provides absolute quantification of brain β-amyloid load which may be valuable for early disease detection and for monitoring the effect of amyloid modifying therapy.
818. Kinetic analysis of a novel radioligand for α7 nicotinic acetylcholine receptor, [11C]CHIBA-1001 in human brain
M. Sakata1, Y. Kimura1,2, M. Ishikawa1,3, J. Toyohara1,4, J. Wu1,4, K. Oda1, K. Ishii1, K. Hashimoto4 and K. Ishiwata1
1Positron Medical Center, Tokyo Metropolitan Institute of Gerontology, Tokyo; 2Molecular Imaging Center, National Institute of Radiological Sciences; 3Department of Psychiatry; 4Center for Forensic Mental Health, Chiba University, Chiba, Japan
Objectives: α7 nicotinic acetylcholine receptor (nAChR) is one of the predominant nAChR subtypes in the brain and is suggested to play an important role in the pathologic states of psychiatric and neurological disorders, such as schizophrenia, Alzheimer's disease, and other dementia. 4-[11C]methylphenyl 2,5-diazabicyclo[3.2.2]nonane-2-carboxylate ([11C]CHIBA-1001) is recently developed as a novel PET tracer for α7 nAChR in the brain.1 Here we performed human PET studies with [11C]CHIBA-1001, and analyzed the kinetics of the tracer in the brain.
Methods: Dynamic PET scans (90-mins) were performed in four normal male volunteers (33±14 ages, non-smoker) on SET-2400W (Shimadzu Co., Kyoto, Japan). The doses of [11C]CHIBA-1001 were 521±107 MBq, and the specific activities were 28.7±9.8 MBq/nmol. Arterial blood was sampled at various time intervals, and the fraction of parent compound in the plasma was determined by HPLC analysis. Eleven ROIs are placed on major cortices, cerebellum, and basal ganglia. ROI-averaged time-activity curves (TACs) were analyzed using one- and two-tissue (1T, 2T) compartment model and Logan graphical analysis (LGA, t* = 30 mins). Total distribution volume (VT) images of [11C]CHIBA-1001 were also estimated by LGA.
VT images of [11C]CHIBA-1001. Total distribution volume images of [11C]CHIBA-1001.
Results: [11C]CHIBA-1001 readily entered the brain. The radioactivity peaked about 15 mins after administration. Metabolism of the [11C]CHIBA-1001 was relatively slow with the percentage of unchanged form in the plasma remaining 73±7% at 60 mins after administration. Figure 1 shows VT images of [11C]CHIBA-1001 estimated by LGA. The tracer was widely distributed in all gray matter regions and no candidate of reference region was found. Regional TACs were well described with 1T model and it caused unstable estimations in 2T model. K1/k2 using 1T model and VT using LGA matched well ((K1/k2) = 1.03 (VT) – 0.3 (r2 = 0.92)). The region which has the highest VT [mL/cm3] was the thalamus (18.2±3.2), followed by the cerebellum (17.8±4.6), amygdala (16.6±3.0), hippocampus (16.5±3.0), posterior cingulate gyrus (16.5±2.1), putamen (16.4±2.8), temporal cortex (16.2±2.5), frontal cortex (15.6±2.4), parietal cortex (15.0±2.4), occipital cortex (14.5±2.2), and caudate (14.5±2.0). Although the distribution of α7 nAChR in all regions of primate brains is still not completely known, the regional distribution pattern of [11C]CHIBA-1001 is consistent with the in vitro results previously reported.2,3
Conclusion: [11C]CHIBA-1001 was suggested to be applicable for further evaluations as a PET tracer for α7 nAChR in human brain.
Acknowledgments: This research was supported by grant from the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation of Japan.
920. Biodistribution study of [61Cu]pyruvaldehyde- bis(N4-methylthiosemicarbazone) in normal rats as a brain PET tracer
A.R. Jalilian1, P. Rowshanfarzad2, F. Bolourinovin1, M. Kamalidehghan1, A. Majdabadi3 and S. Moradkhani1
1Nuclear Medicine Research Group; 2Nuclear Medicine Group; 3Agricultural, Medical and Industrial Research School (AMIRS), Karaj, Iran
Background: [61Cu]-labeled pyruvaldehyde-bis(N4-methylthiosemicarbazone) (61Cu-PTSM) is a promising agent for imaging of blood perfusion that can be prepared in large amounts in the country.
Materials and methods: Copper-61 (T1/2 = 3.33 h) was produced via the natZn(p,x)61Cu nuclear reaction in a 30 MeV cyclotron. 61Cu was separated from the irradiated target by a two-step column chromatography method developed in our laboratory using a cation and an anion exchange resin. The radionuclide was then added to the in-house synthesized PTSM ligand for radiolabeling following the quality control procedures using RTLC and HPLC. The tracer was finaly injected to normal rats and percentages of injected dose per gram was calculated.
Results: After 150 μA irradiation of the target for 76 mins, about 6.006 Ci of 61Cu2+ was obtained with a radiochemical separation yield of more than 95% and a radionuclidic purity of more than 99% (60Cu as impurity). Final 61Cu-PTSM was prepared using the optimized method with a purity more than 98% following administration to normal rats. The tracer is mostly incorporated in heart, kidneys and brain compared to free copper cation as a control which is in agreement with former reports.
Conclusions: [61Cu]-PTSM was prepared at the radiopharmaceutical scales with high quality and is a potential PET tracer in the perfusion study of the heart, kidey, brain and tumors.
985. Microglial activation in leukoaraiosis: a [11C]-PK11195 PET study
A. Bertoldo1, I. Florea2,3,4, V. Di Piero5, A. Panzacchi3,4, M.C. Gilardi2,3,4, G.L. Lenzi5, F. Fazio2,3, C. Cobelli1 and R.M. Moresco2,3,4,6
1Department of Information Engineering, University of Padova, Padova; 2University of Milan ‘Bicocca’; 3San Raffaele Scientific Institute; 4IBFM CNR, Milan; 5University of Rome ‘La Sapienza’, Rome; 6Vita-Salute San Raffaele University, Milan, Italy
Objective: Leukoaraiosis describes diffuse white matter abnormalities on CT or MR brain scans, often seen in the normal elderly and in association with vascular risk factors or stroke.1 The aim of this study was to evaluate by PET [11C]-PK11195 the microglia activation taking into account for both its tissue and vascular expression in patient diagnosed with leukoaraiosis.
Methods: Four healthy controls HC (age 44±11) and two patients with leukoaraiosis (LK), one female (age 64) and one male (age 82) were enrolled in this pilot study. The non invasive simplified reference tissue model2 modified by accounting for cerebral blood volume and vascular binding presence both in reference and target tissues3,4 (SRTMV) was applied to the PET images on a voxel basis to estimate RI (ratio of tissue compared to that in the reference region delivery), k2 (efflux rate constant from tissue), binding potential (BP) and blood volume (Vb). Receptor free regions (Cref) were identify by cluster analysis. The whole blood tracer activity (Cb) was extracted from the dynamic images by averaging the tracer activities in 6 pixels selected by cluster analysis. Cref and Cb were used as input functions to SRTMV, Vb in the reference region was fixed to 5%. Regional BP and Vb values were obtained by drawing regions of interest (ROIs) on BP and Vb parametric images.
Results: The results reveal significant group level effect for both BP and Vb, in cortical and subcortical ROIs. In particular, LK BPs show an increase respect to HC BPs in cortical areas as cuneus, precuneus of about 3 and 3.7 fold, respectively, with a mean fold of 2±0.4 overall the neocortex and in subcortical areas a mean fold increase of about 1.7±0.2. Vb estimates in HC group are >30% lower in cortical area as cuneus, posterior cingulate, insula and parietal cortex. Noteworthy, a variation between groups of more than 20% in Vb values was also identified for occipital cortex, posterior cingulate and thalamus. Average Vb values for HC was 5.7±1% while for LK 7.2±2%.
Conclusions: As observed in patients with AD, Vb correction promotes an increase in BP values in different brain regions. However, differently to what observed for AD patients, in the two subjects with LK, we found Vb values higher than these present in HC group This might be cause by inflammation or modification in peripheral benzodiazepine receptor availability in micro-vessel wall. A blood-brain barrier dysfunction, due to toxic effects of serum protein, and/or the occurrence of ‘incomplete infarction’ have been hypothesized as LK mechanisms, which might be both part of a broader failure of endothelial function. This preliminary study provides new information on the inflammatory process that accompany the LK disease.
1035. Metabolic characterization of 18F-FEOBV, an acetylcholine vesicular transporter ligand, in the rodent
J.-P. Soucy1, É. Landry St-Pierre1, M.-A. Bédard2, P. Rosa1, A. Aliaga1 and G. Massarweh1
1McConnell Brain Imaging Centre, Montreal Neurological Institute, McGill University; 2Neuropsychology, Université du Québec à Montréal, Montreal, QC, Canada
18F-Fluoroethoxy-benzovesamicol (18F-FEOBV) is a high speicificity positron emitting ligand of the acetylcholine (ACh) vesicular transporter, which shows reversible binding kinetics. It holds promise as a potential ACh system pre-synaptic marker which could be useful for early detection of neurodegenerative diseases where ACh neurotransmission is altered such as Alzheimer's, Parkinson's disease or Progressive Supranuclear Paralysis. In order to prepare for its use it in humans, characterizarion of its kinetics and metabolic fate in animals is necessary.
We therefore set up 2 experiments to perform this initial evaluation. In the first one, seven male Sprague-Dawley rats (anaesthetized with isofluorane 2%) were placed in a CTI Concorde R4 microPET scanner. Physiological parameters (respiration rate, EKG and temperature) were recorded throughout the imaging sessions. They received an i.v. dose of 4.3 to 16.7 MBq of high specific activity 18F-FEOBV. Emission scans were obtained for 60 mins. Images were co-registered to a rat brain anatomical (MRI) template. In the second study, five male Sprague-Dawley rats received an average i.v. dose of 37 MBq of high specific activity 18F-FEOBV and were then sacrificed at 5, 7, 10, 24 and 70 mins. Blood was drained and centrifuged and plasma was analyzed for metabolites using reverse-phase HPLC.
Physiological parameters remained constant during the imaging experiment. Rats manifested no overt acute nor subacute (days) signs of toxicity. Distribution of the tracer was found to be as expected from the literature on ACh systems anatomy. There was a fast washout of radioactivity from the cerebellum. On parametric images, the highest binding potentials were detected in the caudate, amygdala, hippocampus and basal forebrain. In the second study, blood analysis showed the presence of a single, hydrophilic, metabolite. The parent compound had a mean retention time of 120 secs with the HPLC set-up we used, whih required extensive fine-tuning do define optimal conditions. Importantly, no lipophilic metabolite was found. Although no physiologic monitoring was done, rats showed no overt sign of physiological distress until sacrifice (up to 70 mins), except for one animal who received a significantly higher mass of the compound.
These results show that 18F-FEOBV is a promising ligand for imaging the innervations density of the cholinergic system, a potentially important parameter in neurodegenerative diseases assessment, which could lead to earlier detection of disease. In the current (and other) experiments, 18F-FEOBV induced no overt toxicity, an encouraging result in terms of its potential clinical and research application in humans. Its metabolism is fairly rapid but as only a hydrophilic metabolite was found, modeling tracer uptake should be straightforward; moreover, the absence of significant uptake in the cerebellum should allow for a non-invasive, Simplified Reference Tissue Analysis type of approach to be used. We are now going through more advanced toxicology testing and are working on optimisation of the tracer imaging and quantification protocol. We will also assess its defluorination in primates before proceeding to human imaging.
1076. The kinetics of FDG uptake in the choroid plexus and the CSF
M. Haddad-Rmeilly1,2, O. Balédent1,2, S. Stoquart-El Sankari1,3, J.-M. Serot4, P. Bailly2 and M.-E. Meyer1,2
1Department of Imaging and Biophysics; 2Department of Nuclear Medicine; 3Department of Neurology; 4Department of Geriatrics, Amiens University Hospital, Amiens Cedex, France
Objectives: Cerebrospinal fluid (CSF) plays a fundamental role in brain pathophysiology. Recent research has underlined the CSF's involvement in neurodegenerative processes, such as Alzheimer's disease (AD). The CSF is produced by the choroid plexus (CP) but the latter's functional activity has only been studied in vitro. we present a new approach for studying these targets with [18F]fluorodeoxy-D-glucose (18F-FDG) positron emission tomography (PET).
Methods: The 45 mins dyn-PET acquisition was scheduled in 34 time frames. Detection was performed in 3D mode. Our novel method provided successive images, representing the FDG uptake dynamics. Firstly, the analysis consisted in calculating 3 parameters (red+green+blue, RGB) for the fitting function (exponential fitting) for the 34 frames. This calculation was performed for every voxel in the brain volume. Secondly, the FDG uptake dynamic of each voxel was visualized as a new colour image generated from the 3 fundamental RGB colour maps. CSF and CP regions of interest were manually selected on the reconstructed map. Mean raw uptake curves were reconstructed.
Results: The Figure shows that inside the right ventricle, one can see a region of interest which appears only in our new parametric images and is absent on the conventional CT and static PET images. In our opinion, this region of interest corresponds to the choroid plexi. The results show for the first time that it is possible to monitor the kinetics of FDG uptake by the PC and the CSF during a PET scan. The FDG kinetics in the PC differ completely from those seen in the CSF and all other brain tissues.
A sample patient image for the same brain slice presented in several difference image modes: the 3 parametric PET maps, the reconstructed PET map, the CT image and a static PET image.
Conclusion: This work is an initial step towards using PET to evaluate CSF production and PC function in vivo.
CristyMEckermanK. ORNL Report ORNL/TM-8381 V1-V7. Oak Ridge, TN, Oak Ridge National Laboratory, 1987.
9.
StabinMGSparksRB. Journal of Nuclear Medicine2005;46:1023–7.
10.
TerryG. Neuroimage2008;41:690–8.
11.
YasunoF. Neuropsychopharmacology2008;33:259–69.
12.
Molecul Pharmacol1988;34:800–5.
13.
Biochem Biophys Res Commun2003;311:847–52.
14.
J Photochem Photobio B: Biol1998;44:29–38.
15.
BergstromM. Neuropharmacology2000;39:664–70.
16.
ZamunerS. Nucl Med Biol2002;29:115–23.
17.
CunninghamV. Int Congress Series2004;1265:12–24.
18.
AmetameySMTreyerV. Human PET studies of metabotropic glutamate receptor subtype 5 with 11C-ABP688. J Nucl Med2007;48:247–52.
19.
HintermannSVranesicI. ABP688, a novel selective and high affinity ligand for the labeling of mGlu5 receptors: Identification, in vitro pharmacology, pharmacokinetic and biodistribution studies. Bioorg Med Chem2007;15:903–14.
20.
PaxinosGWatsonC. The Rat Brain in Stereotaxic Coordinates. Elsevier Academic Press, 2005.
21.
The trial is sponsored and supported by the Bayer-Schering Pharma AG.
22.
HashimotoK. PLoS ONE2008;3:e3231.
23.
CourtJ. Biol Psychiatry2001;49:175–84.
24.
CourtJ. J Neurochem1999;73:1590–7.
25.
McCombePAReadSJ. Int J Stroke2008;3:254–65.
26.
LammertsmaAAHumeSP. Neuroimage1996;3:153–8.
27.
Gunn. Neuroimage1998;8:426–40.
28.
TomasiG. J Nucl Med2008;49:1249–56.
29.
SerotJMBeneMCFoliguetBFaureGC. Morphological alterations of the choroid plexus in late-onset Alzheimer's disease. Acta Neuropathol2000;99:105–8.