
Abstract
We investigated the neuroprotective effect of atorvastatin in combination with delayed thrombolytic therapy in a rat model of embolic stroke. Rats subjected to embolic middle cerebral artery (MCA) occlusion were treated with atorvastatin at 4 h, followed by tissue plasminogen activator (tPA) at 6 or 8 h after stroke. The combination of atorvastatin at 4 h and tPA at 6 h significantly decreased the size of the embolus at the origin of the MCA, improved microvascular patency, and reduced infarct volume, but did not increase the incidence of hemorrhagic transformation compared with vehicle-treated control animals. However, monotherapy with tPA at 6 h increased the incidence of hemorrhagic transformation and failed to reduce infarct volume compared with the control group. In addition, adjuvant treatment with atorvastatin at 4 h and with tPA at 6 h reduced tPA-induced upregulation of protease-activated receptor-1, intercellular adhesion molecule-1, and matrix metalloproteinase-9, and concomitantly reduced cerebral microvascular platelet, neutrophil, and fibrin deposition compared with rats treated with tPA alone at 6 h. In conclusion, a combination of atorvastatin and tPA extended the therapeutic window for stroke to 6 h without increasing the incidence of hemorrhagic transformation. Atorvastatin blocked delayed tPA-potentiated adverse cerebral vascular events, which likely contributes to the neuroprotective effect of the combination therapy.
Introduction
Stroke is a leading cause of morbidity and mortality worldwide. At present, intravenous infusion of tissue plasminogen activator (tPA) is the only FDA (Food and Drug Administration)-approved treatment for acute ischemic stroke (Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995). However, the use of intravenous tPA is limited by its narrow therapeutic window and by the risk of intracerebral hemorrhage (Hacke et al, 2008). An adjuvant agent that enhances the efficacy of thrombolytic therapy without jeopardizing patients with hemorrhagic complication would make the thrombolytic therapy accessible to more stroke patients.
Statins (3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) have pleiotropic effects and are currently considered as potential neuroprotective agents in the treatment of stroke (Endres, 2005). Emerging experimental studies show that statins exert multiple protective effects on cerebral vascular function after acute stroke, i.e., statins are antithrombotic, antiinflammatory, and reduce blood–brain barrier (BBB) disruption (Laufs et al, 2000; Liu et al, 2006; Stuve et al, 2003; Zhang et al, 2005). Therefore, the vascular protective role of acute statin treatment may extend the therapeutic window of thrombolysis for the treatment of stroke. Statins are well tolerated and have been widely used in stroke patients (Collins et al, 2004). A recent pilot-clinical study indicated that treatment with simvastatin initiated at 3 to 12 h from symptom onset improves neurologic outcome (Montaner et al, 2008). Accordingly, this study was undertaken to determine whether acute atorvastatin treatment extends the therapeutic window for tPA in rats after embolic stroke.
Materials and methods
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Henry Ford Hospital.
Animal Model
Male Wistar rats (n=70) weighing 350 to 450 g were subjected to embolic middle cerebral artery (MCA) occlusion (Zhang et al, 1997). Rectal temperature was maintained at 37°C throughout the surgical procedure using a feedback-regulated water heating system.
Experimental Protocols
Atorvastatin (Pfizer, New York, NY, USA) was administered subcutaneously at a dose of 20 mg/kg 4 h after embolic MCA occlusion, followed by a second dose of 20 mg/kg at 24 h after the first dose. The dose of atorvastatin was selected on the basis of the literature, which showed that atorvastatin at a dose of 20 mg/kg is effective in reducing ischemic cell damage in experimental stroke (Laufs et al, 2002; Zhang et al, 2005, 2007). Recombinant human t-PA (tPA, Genentech, San Francisco, CA, USA) was infused intravenously at a dose of 10 mg/kg (10% bolus 6 h after ischemia, and the remainder at a continuous infusion over a 30-min interval using a syringe infusion pump; Harvard Apparatus, Holliston, MA, USA). Owing to elevated plasma levels of plasminogen activator inhibitor 1 (PAI-1), the specific fibrinolytic effect of tPA is at least 10 times lower in rats than in humans (Korninger and Collen, 1981). This dose of tPA (10 mg/kg) is commonly used for investigating the effect of fibrinolysis in rodents (Chopp et al, 1999; Niessen et al, 2002). After embolization, animals were randomly assigned to monotherapy with atorvastatin at 4 h and again at 28 h (n=16), with tPA at 6 h (n=15), saline (n=16), combination of tPA at 6 h and atorvastatin at 4 and 28 h (n=16), or a combination of tPA at 8 h and atorvastatin at 4 and 28 h (n=7). Rats were killed at 30 h or 7 days after MCA. All outcome measurements were performed by observers blinded to treatment.
Functional Outcome
The Modified Neurological Severity Score (mNSS) is a composite of motor, sensory, reflex, and balance tests (Chen et al, 2003). Neurologic function was graded with mNSS at 1 and 168 h after stroke onset.
Histopathologic Studies
Rats were killed 7 days after MCA occlusion, and infarct volume was measured on seven H&E (hematoxylin and eosin)-stained coronal sections, as described previously (Swanson et al, 1990).
Gross hemorrhage, defined as blood evident to the unaided eye on the H&E-stained coronal sections, was evaluated on seven coronal sections for each rat (Zhang et al, 2004). The gross hemorrhage rate is presented as the percentage of animals per group with an identified gross hemorrhage on any coronal section.
Quantitative Measurements of Embolus and Cerebral Vascular Patency
To quantify the change of an embolus, Evans blue-labeled clot, which emits red fluorochrome at the origin of the MCA, was measured 30 h after stroke using a fluorescent microscope (Zhang et al, 2004). Briefly, fluorescence within the right intracranial segment of the internal carotid artery (ICA) and the origin of the MCA was digitized using the MCID (MicroComputer Imaging Device; Imaging Research, Linton, Cambridge, UK). The area of Evans blue (mm2) was calculated by tracing the areas on the computer screen, and a summation of the area of Evans blue from the image is presented as the total area of embolus (Zhang et al, 2004).
To examine the patency of cerebral microvessels, FITC (fluorescein isothiocyanate) dextran (2 × 106 molecular weight, Sigma, St Louis, MO, USA; 50 mg per rat) was administered intravenously to rats 30 h after stroke. Rats were killed 5 mins after FITC-dextran injection. Three coronal sections (100 μm) at the bregma −0.2, −0.8, and −2.8 mm, which encompass the entire territory supplied by the MCA from each rat, were digitized and analyzed using the MCID system, as described previously (Zhang et al, 2004). Data are presented as the numbers of FITC pixels divided by the total numbers of pixels within the field of view, expressed as a percentage.
Immunohistochemistry
Immunostaining was performed on coronal sections obtained from rats that were killed 30 h after MCA occlusion. To examine platelet accumulation, a rabbit polyclonal antibody against rat thrombocyte (Inter-Cell Technologies, Gaithersburg, MD, USA) was used at a titer of 1:4,000. Protease-activated receptor-1 (PAR-1) is a G-protein-coupled receptor that mediates thrombin-induced platelet activation. To detect microvascular PAR-1 expression, a monoclonal anti-human PAR-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used at a titer of 1:500. To examine cerebrovascular fibrin deposition, a goat anti-mouse fibrinogen/fibrin antibody (Accurate Chemical & Scientific, Westbury, NY, USA) was used at a titer of 1:1,000. To examine the integrity of cerebral microvessels, a mouse anti-rat type IV collagen mAb (Abcam, Cambridge, MA, USA) was used at a titer of 1:500. To detect the presence of matrix metalloproteinase-9 (MMP9) in microvessels, a mouse anti-rat MMP9 (95 kDa) antibody (Chemicon, Billerica, MA, USA) was used at a titer of 1:100. To examine cerebral inflammatory responses, a monoclonal anti-rat intercellular adhesion molecule-1 (ICAM-1) antibody (BD, San Jose, CA, USA) was used at a titer of 1:50, and a polyclonal rabbit anti-human MPO (myeloperoxidase) antibody at a titer of 1:500 (DAKO, Carpinteria, CA, USA) was used to examine neutrophil accumulation. For the measurement of vascular fibrin, neutrophil, platelet, ICAM-1, PAR-1, and MMP9 expression, eight nonoverlapping fields of view in the ischemic boundary zone were acquired and digitized using a × 40 objective through the MCID system (Zhang et al, 2004). Numbers of fibrin/fibrinogen, thrombocyte, MPO, and ICAM-1 immunoreactive vessels were counted throughout each field of view. Data are presented as density of immunoreactive vessels within the total scan area (mm2). The area of PAR-1 and MMP9 immunoreactive vessels was calculated and presented as a percentage of the scan area. For semi-quantification of the collagen type IV expression, five fields of view in the territory supplied by the right MCA and five fields of view of the contralateral homologous area were acquired using a × 40 objective through the MCID system. A threshold of color intensity, which encompasses positive vessels on the contralateral image, was applied to each homologous digitized ipsilateral image. Data are presented as a percentage of the positive immunoreactivity area of the contralateral area (pixel).
Statistics
All values are presented as mean±s.e. Two-way ANOVA (analysis of variance) was used to test the synergistic or additive effects of the treatment combination. The analysis began testing for tPA and atorvastatin interaction, followed by group-wise comparison if the interaction or main effect or tPA or atorvastatin was detected at a level of 0.05. If a synergistic effect (a significant interaction) was detected at 0.05, the quantitative synergy ((μ11−μ00)−((μ10−μ00)+(μ01−μ00))) would be estimated with a subadditive effect if (μ11−μ00)−((μ10−μ00)+(μ01−μ00))<0, or with superadditive effects if otherwise. No interaction indicates that combination effects are additive. A subadditive effect indicates that, compared with controls, there was a significant reduction in the combined treated group than an additive reduction in the individually treated group. Logistic regression was used to test gross hemorrhagic rates among the groups.
Results
The Effect of Combination Treatment on Infarct Volume, Incidence of Hemorrhage, and Neurologic Outcome
Treatment with atorvastatin alone at 4 h or with tPA alone at 6 h did not reduce infarct volume compared with the infarct volume in saline-treated rats (Figure 1). However, a combination treatment with atorvastatin at 4 h and tPA at 6 h but not at 8 h significantly (P<0.05) reduced infarct volume compared with that in saline-treated rats (Figure 1). The combination treatments did not increase the incidence of gross hemorrhage (13% in 6 h (1 out of 8) and 14% in 8 h (1 out of 7) groups) compared with that in saline (13%, 1 out of 8), atorvastatin alone (25%, 2 out of 8), and tPA alone (43%, 3 out of 7) groups.
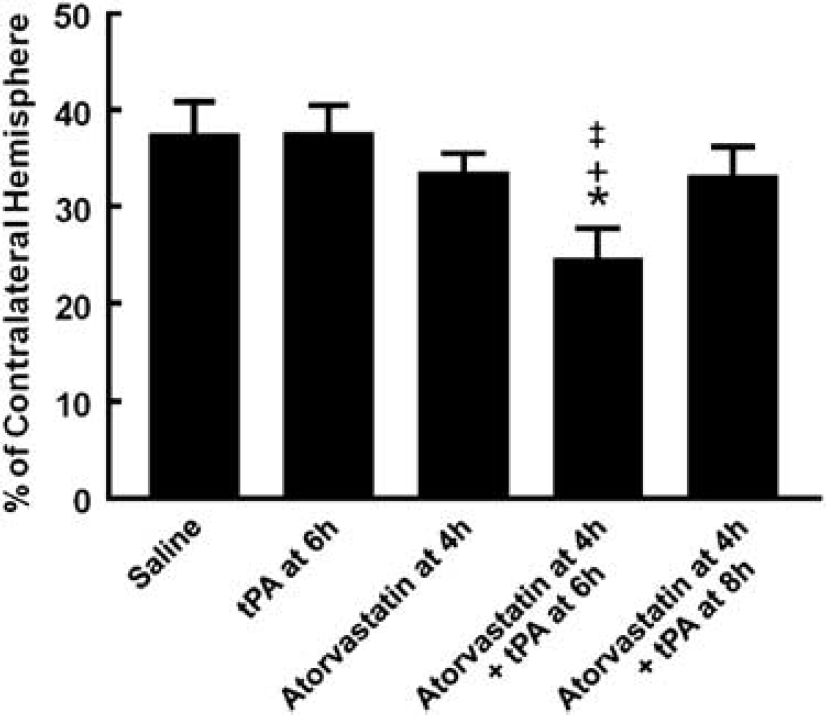
Infarct volume. Bar graph shows the effects of monotherapy with tPA and atorvastatin and the combination therapy on infarct volume assessed 7 days after MCA occlusion. Values are mean±s.e. ∗P<0.05 as compared with that of the saline-treated group.
Neurologic deficits were measured with mNSS 1 h and 7 days after MCA occlusion. One hour after embolic MCA occlusion, all rats showed high scores of mNSS, indicating severe neurologic functional deficits (Table 1). No significant differences were detected among the groups (Table 1). However, 7 days after stroke, the mNSS score in rats treated with a combination of atorvastatin at 4 h and tPA at 6 h was significantly (P=0.007) reduced compared with that in rats treated with atorvastatin alone or with tPA alone (Table 1). Statistical analysis showed that the combination of atorvastatin at 4 h and tPA at 6 h had a synergistic effect on the mNSS score compared with each monotherapy. The combination treatment with atorvastatin at 4 h and tPA at 6 h marginally reduced the mNSS score compared with the saline group (Table 1). Combination treatment with atorvastatin at 4 h and tPA at 8 h failed to reduce the mNSS score compared with saline-treated rats.
Modified neurologic severity score
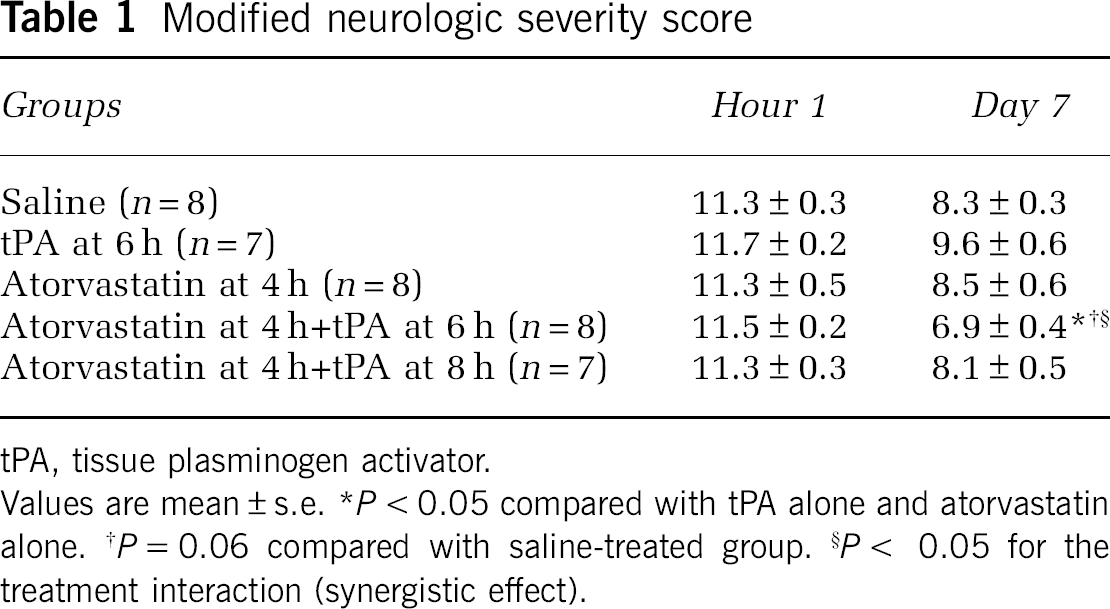
tPA, tissue plasminogen activator.
Values are mean±s.e. ∗P<0.05 compared with tPA alone and atorvastatin alone. †P=0.06 compared with saline-treated group. §P< 0.05 for the treatment interaction (synergistic effect).
The Effect of Combination Treatment on Thrombolysis and Vascular Patency
To examine whether combination treatment promotes thrombolysis, residual clot and thrombosis were measured within the intracranial segment of the right ICA and in parenchymal vessels, respectively, 30 h after MCA occlusion. A large segment of the Evans blue-labeled clot was detected within the intracranial segment of the right ICA and in the origin of the MCA in rats (n=4 per group) treated with saline, atorvastatin alone, or tPA alone (Figure 2). However, rats (n=4) treated with the combination of atorvastatin at 4 h and tPA at 6 h showed a significant (P<0.05) reduction in the area of Evans blue-labeled clot compared with rats treated with saline (Figure 2).
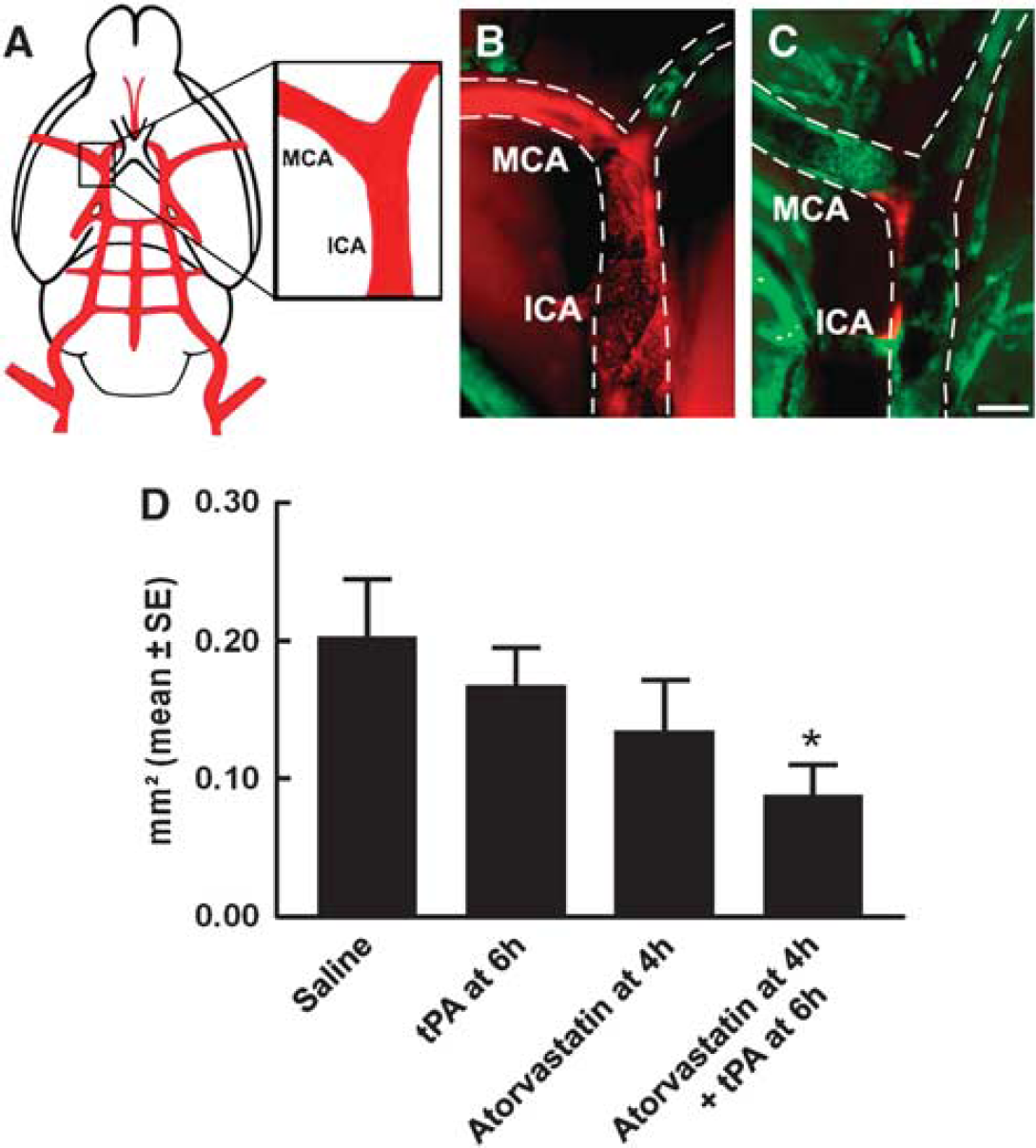
Embolus at the origin of the occluded MCA. (
Treatment with tPA alone 6 h after stroke significantly increased the number of thrombocyte, fibrin/fibrinogen, and MPO immunoreactive cerebral vessels compared with the number in the saline group (Figure 3), indicating an increase in secondary thrombosis. Monotherapy with atorvastatin 4 h after stroke reduced fibrin/fibrinogen deposition by 29% and did not significantly alter platelet deposition within cerebral vessels compared with the saline group (Figure 3). In contrast, the combination treatment with atorvastatin at 4 h and tPA at 6 h significantly reduced the number of thrombocyte, fibrin/fibrinogen, and MPO immunoreactive cerebral vessels compared with the number in the saline, tPA alone, and atorvastatin alone groups (Figure 3). In addition, the combination treatment substantially increased areas perfused by FITC-dextran compared with the saline and tPA alone groups (Figure 3), indicating an enhancement of vascular patency.
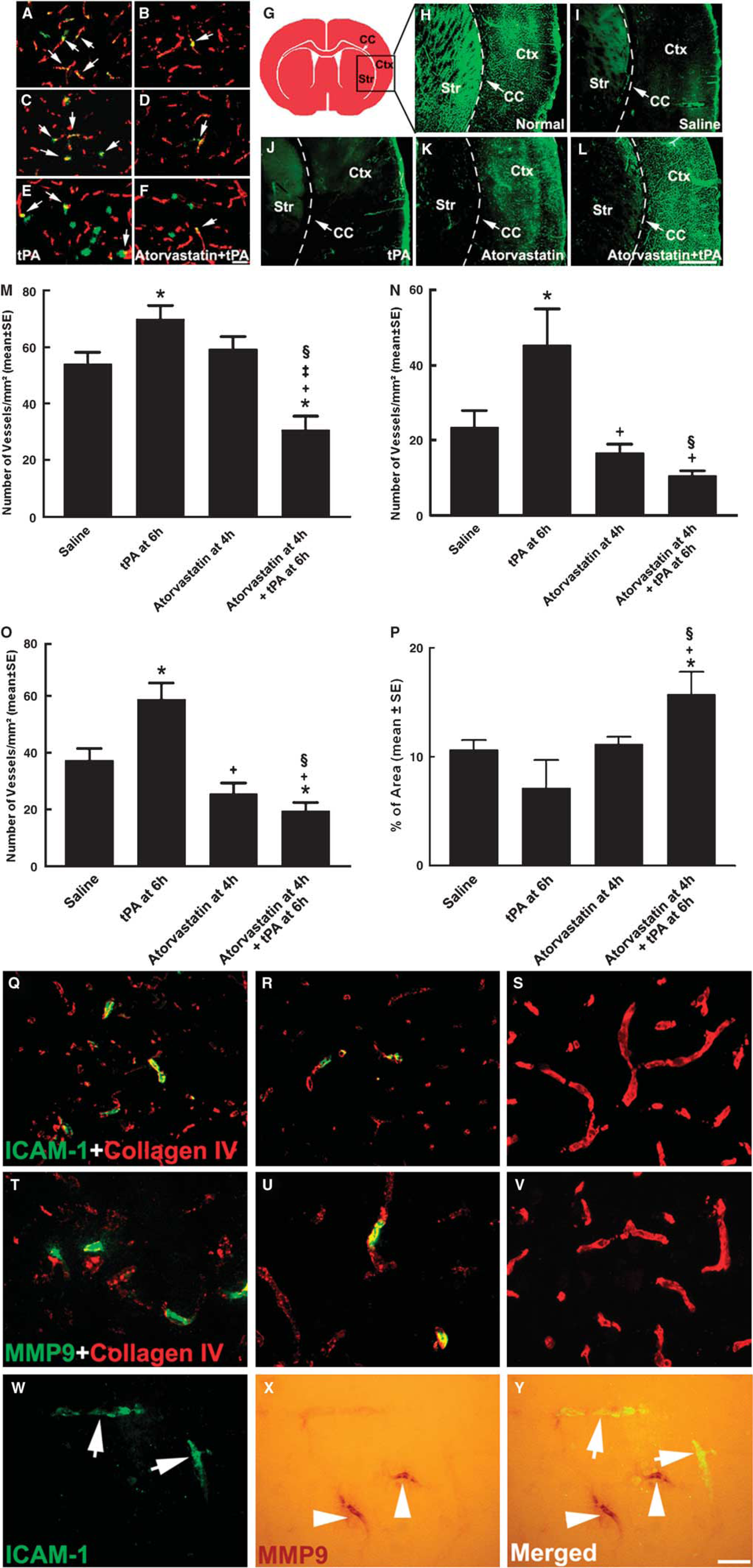
Thrombocyte, fibrin/fibrinogen, MPO, ICAM-1, MMP9, and collagen type IV immunoreactive cerebral vessels and FITC-dextran perfusion. Double immunofluorescent staining (
The Effect of Combination Treatment on Activation of Endothelial Cells
Delayed thrombolysis with tPA triggers the procoagulation of gene expression in activated cerebral endothelial cells (Liu et al, 2006). Therefore, quantitative measurements of ICAM-1 and PAR-1 immunoreactive vessels were performed 30 h after MCA occlusion. Monotherapy with tPA 6 h after stroke but not with atorvastatin significantly increased the number of ICAM-1 and PAR-I immunoreactive vessels compared with the number in the saline-treated rats (Table 2). Interestingly, monotherapy with atorvastatin reduced ICAM-1 immunoreactive vessels by 22% compared with the number in the saline group, although the reduction did not reach statistical significance (Table 2). However, combination treatment with atorvastatin at 4 h and tPA at 6 h significantly reduced the number of ICAM-1 and PAR-1 immunoreactive vessels (Table 2), suggesting that the combination therapy suppresses the procoagulation of gene expression.
Vascular ICAM-1, PAR-1, collagen type IV, and MMP9
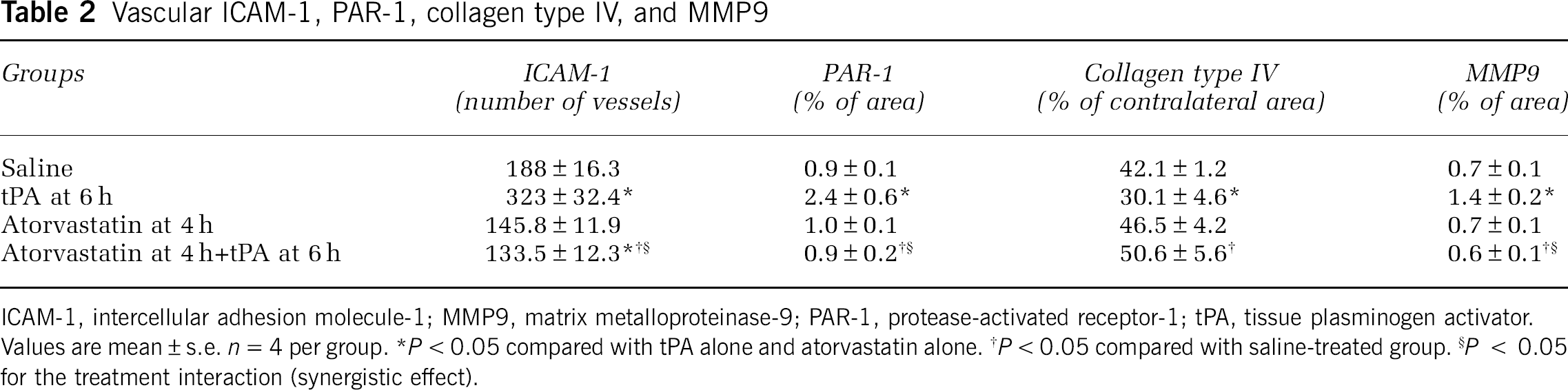
ICAM-1, intercellular adhesion molecule-1; MMP9, matrix metalloproteinase-9; PAR-1, protease-activated receptor-1; tPA, tissue plasminogen activator.
Values are mean±s.e. n=4 per group. ∗P<0.05 compared with tPA alone and atorvastatin alone. †P<0.05 compared with saline-treated group. §P < 0.05 for the treatment interaction (synergistic effect).
The Effect of Combination Treatment on Cerebrovascular Integrity
Collagen type IV is an important constituent of the BBB (Hamann et al, 1995). MMP9 degrades collagen type IV and mediates BBB breakdown after stroke (Asahi et al, 2001). Quantitative measurements of collagen type IV and MMP9 immunoreactive vessels 30 h after stroke showed that combination treatment with atorvastatin at 4 h and tPA at 6 h significantly preserved the collagen type IV immunoreactive area, which was associated with a significant reduction in MMP9 immunoreactive vessels at the ipsilateral hemisphere compared with saline-treated rats (Table 2). Conversely, treatment with tPA alone 6 h after stroke significantly reduced the collagen type IV immunoreactive area and increased numbers of MMP9 immunoreactive vessels compared with saline-treated rats (Table 2). Atorvastatin alone neither reduced collagen IV loss nor increased the number of MMP9-positive vessels compared with saline-treated rats (Table 2).
Double immunostaining showed that cerebral vessels with diffused collagen type IV immunoreactivity were MMP9 or ICAM-1 positive (Figure 3), whereas MMP9-positive vessels were not colocalized to ICAM-1 immunoreactivity (Figure 3), suggesting that both ICAM-1 and MMP9 contribute to loss of microvascular integrity.
Discussion
This study shows that combination of atorvastatin at 4 h and tPA at 6 h significantly reduces infarct volume and improves neurologic function without increasing the incidence of hemorrhage in the rat after embolic stroke. These data suggest that atorvastatin blocks tPA-potentiated adverse effects on cerebral vascular patency and integrity, thereby extending the therapeutic window of thrombolysis with tPA to 6 h.
When a single embolus is placed at the origin of the MCA, the contents of the embolus at the origin of the MCA change with time from a fibrin-rich clot to a composite of fibrin, platelets, and leukocytes, which are resistant to fibrinolysis (Braaten et al, 1994; Reed et al, 1992; Zhang et al, 2001). Occlusion of the MCA also triggers thrombosis in downstream microvessels (Zhang et al, 2001). These findings show that the combination of atorvastatin (4 h) and tPA (6 h) significantly reduced clot size at the origin of the MCA and increased downstream microvascular patency, indicating that combination therapy promotes recanalization. Immunohistochemistry analysis in this study showed that monotherapy of tPA 6 h after stroke significantly increased fibrin, platelet, and neutrophil deposition within cerebral vessels compared with the saline group. In addition, tPA monotherapy upregulated PAR-1 and ICAM-1 molecules, which are known to mediate the formation of thrombosis (Coughlin, 2000; Liu et al, 2006; Zhang et al, 1999). These data are consistent with published studies that showed that delayed treatment with tPA increases secondary thrombosis (Fitzgerald et al, 1988; Liu et al, 2006). In contrast to tPA, monotherapy of atorvastatin 4 h after stroke reduced fibrin, platelet, and neutrophil deposition within cerebral vessels compared with the saline group, although the reduction did not reach statistical significance. However, the combination therapy of 4 h atorvastatin and 6 h tPA had a synergistic effect on suppressing secondary thrombosis triggered by tPA. It is interesting to note that atorvastatin has been shown to upregulate endogenous tPA and to promote thrombolysis after stroke (Asahi et al, 2005). Therefore, the prothrombolytic effect of the combination therapy likely results from an atorvastatin-mediated reduction of secondary thrombosis, which leads to an early recanalization and a reduction in ischemic lesion volume even when tPA is administered 6 h after stroke. Our data are consistent with published studies showing that statins confer neuroprotection through several mechanisms, including antithrombotic and antiinflammatory actions in experimental stroke (Liu et al, 2006; Vaughan, 2003; Zhang et al, 2005, 2007). Interestingly, the DEFUSE (Diffusion and Perfusion Imaging Evaluation for Understanding Stroke Evolution) study recently showed an association between early recanalization visualized using magnetic resonance angiography and a favorable clinical response in patients with a mismatch between baseline perfusion- and diffusion-weighted imaging findings when patients are treated with intravenous tPA in a 3- to 6-h window (Marks et al, 2008).
Intracerebral hemorrhage is the major complication of thrombolytic therapy in acute stroke (Clark et al, 1999; Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995). Thrombolysis with tPA upregulates MMP9, which degrades the extracellular matrix components including collagen IV, thereby aggravating BBB disruption and hemorrhagic transformation (Lapchak et al, 2000; Sumii and Lo, 2002). In parallel, our data show that monotherapy with tPA 6 h after stroke significantly increased microvascular levels of MMP9, which was associated with a reduction of vascular collagen IV, indicating a disruption of the BBB. However, the combination therapy of atorvastatin and tPA abolished tPA-induced upregulation of MMP9, as well as the degradation of collagen IV, leading to a reduction in the incidence of hemorrhagic transformation (10%) compared with the incidence during monotherapy of the tPA group (40%). Elevation of the MMP9 serum level is associated with hemorrhage transformation in patients with and without thrombolysis treatment (Castellanos et al, 2003; Saqqur et al, 2008). Statins reduce MMP9 activity (Koh et al, 2002). Thus, our data suggest that atorvastatin ameliorates BBB disruption exacerbated by delayed tPA treatment, thereby reducing the incidence of hemorrhagic transformation.
Hemorrhage transformation induced by tPA is also associated with recanalization time (Saqqur et al, 2008). Using this model, we previously showed that treatment with tPA 1 h after stroke significantly reduces infarct volume and increases reperfusion without augmentation of hemorrhage transformation (10%) (Zhang et al, 1998). In contrast, this study shows that intravenous administration of tPA at 6 h did not induce recanalization and increased the incidence of hemorrhage transformation (40%); although, because of sample size, this study was not sufficiently powered to detect statistical significance. Thrombolysis promoted by the combination therapy of atorvastatin (4 h) and tPA (6 h) also contributed to a reduction in hemorrhage transformation. A recent clinical study shows that the risk of tPA-related symptomatic intracerebral hemorrhage is low after an early and complete restoration of blood flow (Saqqur et al, 2008).
We and others have shown that acute treatment of stroke with atorvastatin upregulates the PI3K (phosphoinositide 3-kinase)/Akt signaling pathway and endothelial NOS (eNOS), which likely are the underlying mechanisms that contribute to the extension of the thrombolytic window for stroke (Laufs et al, 2000; Liu et al, 2006; Zhang et al, 2007). Atorvastatin attenuates the procoagulation of gene expression triggered by tPA in cerebral endothelial cells, whereas blockage of the PI3K/Akt pathway abolishes these effects (Liu et al, 2006; Zhang et al, 2007). In this study, atorvastatin downregulates tPA-potentiated vascular thrombogenic events and ameliorates BBB disruption. Treatment of stroke with atorvastatin upregulates eNOS, which leads to an increased cerebral blood flow (Endres et al, 1998). Thus, we speculate that atorvastatin activates the PI3K/Akt pathway and/or eNOS, which suppresses multiple downstream genes involving the formation of secondary thrombosis and disruption of the BBB, as well as increases cerebral blood flow after stroke and tPA treatment. Consequently, these processes contribute to neuroprotection.
The dose of atorvastatin used in this study has been shown to reduce ischemic cell damage and does not significantly alter the mean arterial blood pressure and total cholesterol levels after stroke in the rat (Zhang et al, 2005). Administration of atorvastatin 20 mg/kg once daily for 20 days does not induce toxicity in rats (Black et al, 1999). Clinical data suggest that a high dose of atorvastatin is more effective in reducing inflammatory reaction than a low dose in patients with acute coronary syndromes or diabetes (Kinlay et al, 2003; van de Ree et al, 2003). Therefore, adjuvant treatment with atorvastatin may be a valuable approach to extend the therapeutic time window of tPA for the treatment of stroke. Although statins are a class of drugs that exert vasoprotective effects after stroke, different statins may have different noncholesterol effects (Rosenson and Tangney, 1998; Turner et al, 2007). Whether combination of tPA with statins other than atorvastatin will have the same neuroprotective effects observed in this study remains to be determined. Another potential caveat of our study is that the concentration of tPA used in this study is 10-fold higher than the clinical dose. Despite the differences in the fibrinolytic activity between humans and rodents, the relatively high dose of tPA may directly trigger proinflammation and procoagulation of genes in cerebral vasculature, and exert multiple tissue toxic effects. Therefore, our results on tPA-potentiated vascular adverse effects should be interpreted with caution.
In conclusion, adjuvant treatment with atorvastatin increases the therapeutic window for tPA to 6 h after stroke. Neuroprotection of the combination therapy is presumably due to the effect of atorvastatin on thrombolytic efficacy, which leads to cerebral vascular patency and integrity.
Footnotes
The authors declare no conflict of interest.