
Abstract
Recent studies have shown the influence of glucocorticoids on the expression of the tight junction protein occludin in the brain capillary endothelial cell line cEND, contributing to improvement in endothelial barrier functions. In this study, we investigated glucocorticoid effects on the expression of the adherens junction proteins VE- (vascular-endothelial) cadherin, α-catenin and β-catenin as well as that of ZO-1, the plaque protein shared by both adherens and tight junctions on stimulation with dexamethasone. We were able to show a positive influence of dexamethasone administration on VE-cadherin protein levels as well as a rearrangement of VE-cadherin protein to the cytoskeleton after dexamethasone treatment. Investigation of transcriptional activation of the VE-cadherin promoter by dexamethasone, however, did not point to direct glucocorticoid-mediated VE-cadherin gene induction but rather suggested indirect steroid effects leading to increased VE-cadherin protein synthesis. Dexamethasone was further shown to induce cellular differentiation into a cobblestone cellular morphology and reinforcement of adherens junctions concomitant with the increased anchorage of VE-cadherin to the actin cytoskeleton. We thus propose that glucocorticoid effects on VE-cadherin protein synthesis and organization are important for the formation of both adherens and tight junction, and for improved barrier properties in microvascular brain endothelial cells.
Introduction
The cadherin superfamily includes calcium-dependent single-pass adhesion molecules controlling cell architecture, cell—cell recognition, and survival (Gumbiner, 1996). Cadherins are frequently associated with characteristic junctional structures called adherens junctions (AJs) (Gumbiner, 2005). They build homotypic dimers through the extracellular domain that adhere to dimers of the opposing cell membrane and tend to form clusters at the cell surface, which strengthen intercellular adhesion (Baumgartner et al, 2003). The cytoplasmic tail of classic cadherins interacts in the junctional complex with β-catenin and placoglobin/γ-catenin, which in turn binds to α-catenin (Perez-Moreno et al, 1998). The latter is able to anchor actin filaments in a direct manner or indirectly through actin binding proteins such as vinculin or α-actinin (Perez-Moreno et al, 1998). Cadherins are expressed in a tissue-specific manner: VE- (vascular-endothelial) cadherin is characteristically expressed in the endothelium (Gumbiner, 1996) and it is one of the first markers detected in endothelial progenitor cells (Vittet et al, 1996), indicating that it has an important role in development. It contributes to maintenance of cell morphology and control of cell motility through binding of the actin-based cytoskeleton (Gumbiner, 1996). VE-cadherin is strongly expressed in peripheral and neural endothelial cells (ECs), where it appears to be the major cell—cell adhesion molecule. Consequently, it is generally thought that formation of a paracellular barrier requires the initial engagement of cell—cell contacts at the AJ, before the tight junction (TJ) can be established to form a continuous seal around the lateral circumference of adjacent cells (Gumbiner, 1996; Madara, 1998). In the course of establishment of the junctional complex, a series of molecular processes then appear to trigger the recruitment of TJ components to the points of cell—cell contact and formation of a tight paracellular seal of the endothelium dependent on prior establishment of the AJ (Madara, 1998).
Brain edema, tumors, and autoimmune neuroinflammation like multiple sclerosis are known to compromise the integrity of the blood—brain barrier (BBB). There are various processes underlying the pathology of these diseases, including monocyte migration across the BBB, phagocytosis and degradation of myelin, axonal degradation, and oligodendrocyte damage. Therapeutical strategies to reconstitute the compromised BBB in these diseases include glucocorticoid (GC) treatment (Engelhardt, 2000), but the molecular mechanisms by which GCs regulate BBB permeability are still poorly understood. Among others, GC action could be described as a repression of proinflammatory transcription factors, for example, nuclear factor-κB (Pitzalis et al, 2002), causing the inhibition of cytokine-induced barrier breakdown and expression of cell adhesion molecules, which mediate T-cell—BBB interactions and excessive leukocyte recruitment across the BBB. Beyond these findings, our observation of GC-mediated induction of the TJ protein occludin in cEND cells generated from immortalized murine brain capillary ECs (Förster et al, 2005) pointed out a new way for understanding the beneficial effects of GC action in a therapeutic regime: GCs are known to act through the glucocorticoid receptor (GR) forming a complex with DNA in the nucleus and consequently modulating gene expression or to mediate nontranscriptional regulation processes by the GR by means of signalling cascades. In our previous works, we were able to show the upregulation of occludin, a component of the TJ complex, strongly expressed in ECs of the BBB as a consequence of genomic GC action (Förster et al, 2005, 2006). Glucocorticoid action also led to a decrease in permeability caused by GC treatment in the cEND cell line (Förster et al, 2005). In the present study, we were able to show an increase in VE-cadherin protein levels in cEND cells as well as a rearrangement of VE-cadherin to the cytoskeleton on GC treatment as a result of late non-genomic GC effects. As another indirect effect of GC treatment, we detected morphologic changes in GC-treated cells, such as reorganization of the actin cytoskeleton into a cortical adhesion belt, concomitant with the strong anchorage of VE-cadherin protein to the cytoskeletal fraction.
Materials and methods
Chemicals
Dexamethasone, hydrocortisone, and mifepristone were purchased from Sigma (Taufkirchen, Germany).
Animals and Collection of Tissues
Neonatal mice (strain 129Sv) of either sex (3 days old) were killed by CO2 asphyxiation. The brains were removed immediately and transferred to a dissection chamber containing buffer A (15 mmol/L HEPES (pH 7.4), 153 mmol/L NaCl, 5.6 mmol/L KCl, 2.3 mmol/L CaCl2·2H2O, 2.6 mmol/L MgCl2·6H2O, 1% (w/v) bovine serum albumin). All the experiments were approved by the local Animal Care Committee (Tierschutzbeauftragter).
Isolation and Culture of Cerebral Endothelial Cells
The immortalized mouse brain capillary EC line cEND was generated as described (Förster et al, 2005). Briefly, brains (cerebrum without cerebellum and brain stem) were isolated from neonatal mice (3 days post partum) as described above, and after removal of the meninges and capillary fragments, the tissue was minced in buffer A using a sterile cutter. Fragments were digested in 0.75% (w/v) collagenase A (Roche, Mannheim, Germany) for 30 mins at 37°C in a water bath (with occasional shaking). Digestion was stopped by the addition of 10 volume of ice-cold buffer A. To remove myelin, centrifugation through a 25% (w/v) bovine serum albumin gradient was carried out for 20 mins at 1,000g. The resulting EC pellet was washed twice with buffer A to remove myelin and bovine serum albumin. Additionally, another 0.75% collagenase A digestion was performed to purify ECs from astrocytes. Primary cells were then resuspended in Dulbecco's modified Eagle's medium (Sigma) supplemented with growth medium (10% heat-inactivated fetal calf serum (FCS)) and 1% (w/v)
Cell Cultures
cEND cells were cultured as described above. The cultures were supplemented with 100 IU/mL penicillin and 100 mg/mL streptomycin (1% PEST). Cells were maintained in an atmosphere of 5% CO2 and at 37°C. HEK293 cells were cultured in Eagle's minimum essential medium (Sigma) with supplements and conditions as described above for cEND cells.
Transendothelial Electrical Resistance
Cells were plated on top of collagen IV-coated transwell chambers of six-well plates (0.4 μm pores) (Falcon, Heidelberg, Germany) at densities of 1 × 104 cells/well. When they reached confluence at day 5, the different experimental sets of cells were treated as indicated in the figure legends. The control set was maintained in basal medium (Dulbecco's modified Eagle's medium, 2% FCS) for an additional 72 h at which point the transendothelial electrical resistance (TER) measurement assays were performed. Transendothelial electrical resistance was measured using an assembly containing current-passing and voltage-measuring electrodes (World-Precision Instruments Inc., New Haven, CT, USA). Resistances of blank filters were subtracted from those of filters with cells before final resistances (in Ω cm2) were calculated. All the experiments were repeated at least four times.
Electrophoresis and Immunoblotting
Cells were plated at a density of 1.1 × 105 cells/3.5 cm2 dish and grown to confluence. At confluence, cells were maintained in 2% FCS and treated with dexamethasone as indicated in the figure legends. cEND cells were thereafter dissolved in Laemmli sample buffer and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, 15% gels). Protein contents were quantified directly from SDS-PAGE loading buffer using 0.1% (w/v) amidoschwarz (AppliChem, Darmstadt, Germany) in 25% (v/v) methanol/5% (v/v) acetic acid. For immunoblotting, proteins were transferred in Kyhse-Andersen transfer buffer (Kyhse-Andersen, 1984) to Hybond nitrocellulose membranes (Amersham, Braunschweig, Germany) that were blocked with 10% (w/v) low-fat milk in phosphate-buffered saline (PBS, pH 7.4, consisting of 137 mmol/L NaCl, 2.7 mmol/L KCl, 8.1 mmol/L Na2HPO4, and 1.5 KH2PO4) and incubated overnight at 4°C with the rat monoclonal antibody 11D4.1 (undiluted hybridoma supernatant) directed to the ectodomain of mouse VE-cadherin (Gotsch et al, 1997). As secondary antibody we used horseradish peroxidase-labelled goat anti-rat IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), diluted 1:3,000 in PBS. α-Catenin and β-catenin were visualized with the rabbit polyclonal antibodies sc-7094 from Santa Cruz Biotechnologies (Santa Cruz, CA, USA) and C-2206 from Sigma, respectively, and for ZO-1 we used rabbit polyclonal antibody from Zymed Laboratories (South San Francisco, CA, USA). As secondary antibody, horseradish peroxidase-labelled goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories) was used at 1:3,000 dilution with PBS. Bound immunoglobulins were visualized by the enhanced chemiluminescence technique (ECL, Amersham). Densitometric analysis using Scion Image Beta 4.02 (Scion Corp., Frederick, MD, USA) was performed for quantification. As a control for equal loading, β-actin western blot (Sigma, A-5441, mouse monoclonal, used at 1:500 dilution) was performed.
Semiquantitative reverse transcription-PCR
Total RNA was extracted from confluent cEND monolayers after 24 and 48 h treatment with dexamethasone or dexamethasone and the GR antagonist RU486, as indicated in the legend to Figure 2B, using the RNeasy Reagent (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Reverse transcription of 2 μg total RNA into cDNA was performed using reverse transcriptase (iSCRIPT cDNA synthesis kit, Bio-Rad, München, Germany) at 25°C for 10 mins and 37°C for 2 h and resulted in 100 μg cDNA. The pair of primers used for amplification of VE-cadherin sequences was VE-cadherin forward (5′-TTGCCCAGCCCTACGAACCTAAAG-3′) and VE-cadherin reverse (5′-ACCACCGCCCTCCTCATCGTAAGT-3′) in a final volume of 50 μL, using 10 mmol/L dNTP (Gibco, Karlsruhe, Germany) and 30 pmol of each primer (MWG Biotech, Ebersberg, Germany). The thermocycler was programmed to give an initial cycle consisting of denaturation at 94°C for 1 min and annealing/extension at 60°C for 30 secs and 72°C for 1 min. To control the amount of total RNA, a parallel reverse transcription-PCR (RT-PCR) of glyceraldehyde phosphate dehydrogenase (GAPDH) as housekeeping gene using the following pair of primers for amplification was performed: GAPDH forward (5′-CAAGACGGACCAGAGCGAAAGC-3′) and GAPDH reverse (5′-CAATCTCGGGTGGCTGAACGC-3′). Both PCR reactions were performed in a mastercycler gradient thermocycler (Eppendorf). PCR products were subjected to electrophoresis on a 1.5% agarose gel and then stained with ethidium bromide. A 100 bp DNA ladder (Generuler, Fermentas, St Leon-Rot, Germany) was used for calibration. Band densities were compared under UV-induced fluorescence. Densitometric analysis using Scion Image Beta 4.02 (Scion Corp.) was performed for quantification.
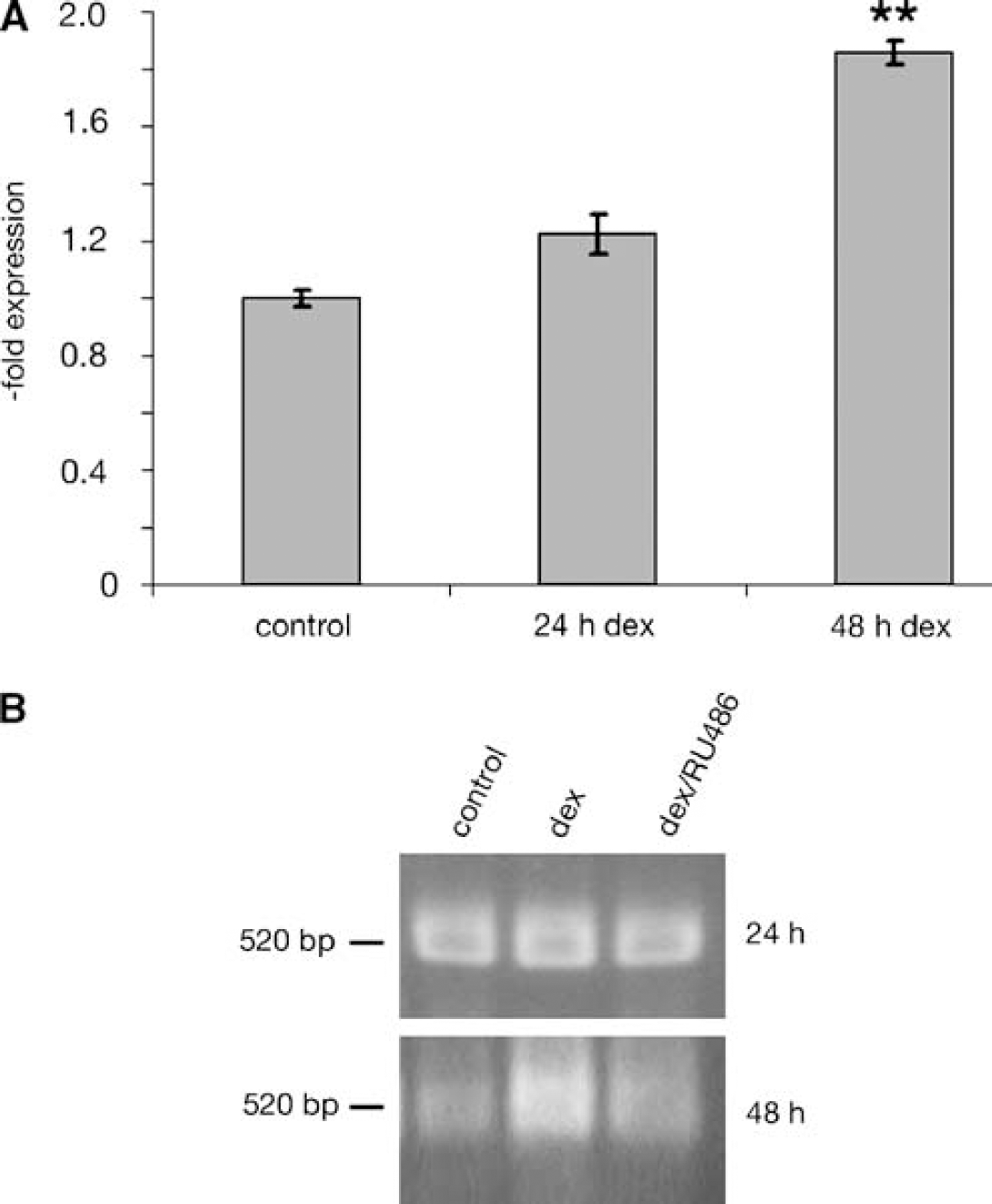
Dexamethasone effects on VE-cadherin gene transcription levels in cEND brain capillary ECs. (
Quantitative Real-Time Reverse Transcription-PCR
For real-time RT-PCR, cDNA was synthesized using the iSCRIPT cDNA synthesis kit (Bio-Rad) and 1 μg of RNA from cEND cells treated or not treated with dexamethasone and harvested at time points indicated in the figure legends. Primers were designed using the Primer Express Software (Applied Biosystems, Foster City, CA) and obtained from MWG Biotech. Real-time RT-PCR was performed using the TaqMan PCR Master Mix (Applied Biosystems) and the TaqMan Gene Expression Assays (no. Mm00486938_m1 for VE-cadherin and no. Mm99999915_g1 for GAPDH). All PCR reactions were performed in triplicate for each target. Data were acquired with the ABI PRISM 7300 system (Applied Biosystems). The ABI PRISM 7300 SDS software (relative quantification study) was used to determine the threshold cycle (Ct) for each reaction and gene expression was normalized to the expression of the endogenous housekeeping gene GAPDH based on the 2−ΔΔCt method.
Transfection and Luciferase Assay
Transfection and luciferase assays were carried out essentially as described previously (Förster et al, 2005). Transient transfection experiments using the Effectene reagent (Qiagen) were performed as described by the manufacturer, using 2 μg of VE-cadherin luciferase reporter vector −2486LUC (in brief −2486LUC; Lelievre et al, 2000), and 1 μg of pTRL-TK (Promega, Madison, WI, USA) in the presence or absence of ligands (as indicated in the figure legends). HEK293 cells were seeded on six-well cell culture plates 24 h before transfection in Eagle's minimum essential medium containing 10% FCS and 1% PEST at a density of 2 × 106 cells/well. cEND cells were seeded in Dulbecco's modified Eagle's medium containing 10% FCS on six-well cell culture plates coated with collagen IV and grown to confluence before transfection.
To assess GC effects on −2486LUC promoter transactivation, after the addition of the DNA/Effectene mixture, cells of either cell line were incubated overnight at 37°C and 5% CO2. After this, 2 mL of fresh medium containing 2% dextran-coated charcoal (DCC) (Fagart et al, 1998)-treated FCS/1% PEST and ligands or vehicle alone (as indicated in the figure legends) was added. After 24 h, cells were washed once with PBS and harvested with 250 μL of passive lysis buffer for active cell lysis. Following this, cellular extracts were prepared according to the manufacturer's instructions. Protein concentration was estimated by standard Bradford protein assay (Bradford, 1976). Cell lysates were then analyzed for luciferase and Renilla activity: measurement of both firefly and Renilla luciferase (de Wet et al, 1985; Wood et al, 1984) activity was performed with the Dual-Luciferase assay kit (Promega) according to the manufacturer's instructions. For this, 20 μL of cell lysate was used for assaying the enzymatic activities, using an LB9507 luminometer with dual injector (Berthold, Bad Wildbad, Germany). Each lysate was measured twice. Promoter activities were expressed as relative light units normalized for the protein content and the activity of Renilla luciferase in each extract. The data were calculated as the mean of five identical setups.
VE-Cadherin Cytoskeletal Association
cEND cells were plated on 25 cm2 culture flasks, switched to low serum (2% (w/v) FCS) medium after reaching confluence, and later stimulated with 100 nmol/L dexamethasone for 72 h every 24 h. Cells were subsequently fractionated with cytoskeleton-stabilizing buffer containing 10 mmol/L KCl, 1 mmol/L EGTA, 3 mmol/L MgCl2, 1 × protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), 1 mmol/L Na3VO4, and 0.5% Triton X-100 by centrifugation at 15,000g for 15 mins. A 50 μL portion of Laemmli sample buffer was added to the Triton X-100-insoluble fraction, which contains cytoskeleton-associated proteins, and incubated at 95°C for 15 mins. Protein contents of both cell fractions were quantified by amidoschwarz method (as described above). Both Triton X-100-insoluble and Triton X-100-soluble fractions were subjected to SDS-PAGE followed by immunoblotting with the rat monoclonal antibody 11D4.1 (undiluted hybridoma supernatant) anti-mouse VE-cadherin (Gotsch et al, 1997).
Immunocytochemistry
Cultured cEND cells were grown on collagen IV-coated 35-mm-diameter coverslips (Marienfeld, Lauda-Königshofen, Germany). After stimulation with 100 nmol/L dexamethasone for 72 h every 24 h, cells were fixed with PBS containing 2% formaldehyde (freshly prepared from paraformaldehyde) for 10 mins at room temperature, washed with PBS, and permeabilized with 0.1% (v/v) Triton X-100 for 5 mins. Cells were blocked with bovine serum albumin/normal goat serum for 30 mins at room temperature and stained with the rat monoclonal antibody 11D4.1 (undiluted hybridoma supernatant) anti-mouse VE-cadherin (Gotsch et al, 1997), used also for immunoblotting experiments, overnight. VE-cadherin reacting with antibody was visualized with Cy3-labelled goat anti-rat IgG, diluted 1:600 in PBS (Dianova, Hamburg, Germany). F-actin could be detected using Alexa-phalloidin (Mobitech, Göttingen, Germany), diluted 1:60 in PBS (incubation for 1 h at room temperature). After the staining procedure, cells were rinsed three times for 5 mins with PBS and coverslips were mounted on glass slides with 60% glycerol in PBS containing 1.5% of the antifading compound n-propyl gallate (Serva, Heidelberg, Germany). Images were recorded by fluorescence microscopy (Axioskop2 plus, Zeiss, Germany).
Analysis and Statistics
Values for densitometry, gene expression, and promoter transactivation were averaged to establish a single value for control cells and treated cells as indicated. Throughout the experiments, averaged values were reported as means±standard deviation (s.d.). Mann—Whitney U-test) was performed and P<0.05 (∗) was considered statistically significant and P<0.001 (∗∗) highly statistically significant.
Results
Having proved occludin as a direct target for GC-mediated improvement of BBB function in brain microvascular cEND cells (Förster et al, 2005, 2006), we intended to test for further potential GC targets among components of the apical junctional complex: the endothelial barrier function appears to be largely dependent on functional AJ and TJ and their linkage to the actin cytoskeleton forming a dense peripheral band that is found at the cytoplasmic face along the junctional borders of adjacent ECs in situ and in highly confluent endothelial cultures (Schnittler, 1998). We thus examined GC effects on cellular morphology and the organization of the actin cytoskeleton.
Dexamethasone Induces Morphologic Changes in cEND Cells through Remodelling of the Cytoskeleton
As GC treatment has been reported to induce morphologic changes in brain microvascular ECs (Förster et al, 2005; Romero et al, 2003), we analyzed changes in cellular morphology, that is, in the organization of the F-actin cytoskeleton, in dexamethasone-treated cEND cells. After 72 h of dexamethasone supplement in the differentiation medium (treatment repeated every 24 h), immunocytochemical analysis revealed a morphologic rearrangement of F-actin in dexamethasone-treated cEND cells compared with untreated cells. This was visualized consistently in dexamethasone-treated cells by the formation of a continuous ring of actin in the form of a cortical adhesion belt (Schnittler, 1998) at the cell borders (Figure 1B) as opposed to actin stress fibers in untreated cells (Figure 1A). Stress fibers were reduced or not detectable in dexamethasone-treated cells (Figure 1B). We observed changes in cellular morphology of treated cEND, which changed their spindle-like elongated shape (Figure 1C) into a more cobblestone-like phenotype (Figure 1D), as observed previously for hydrocortisone-treated cEND cells (Förster et al, 2005) and in primary brain capillary ECs and epithelial cells (Lossinsky and Shivers, 2004).
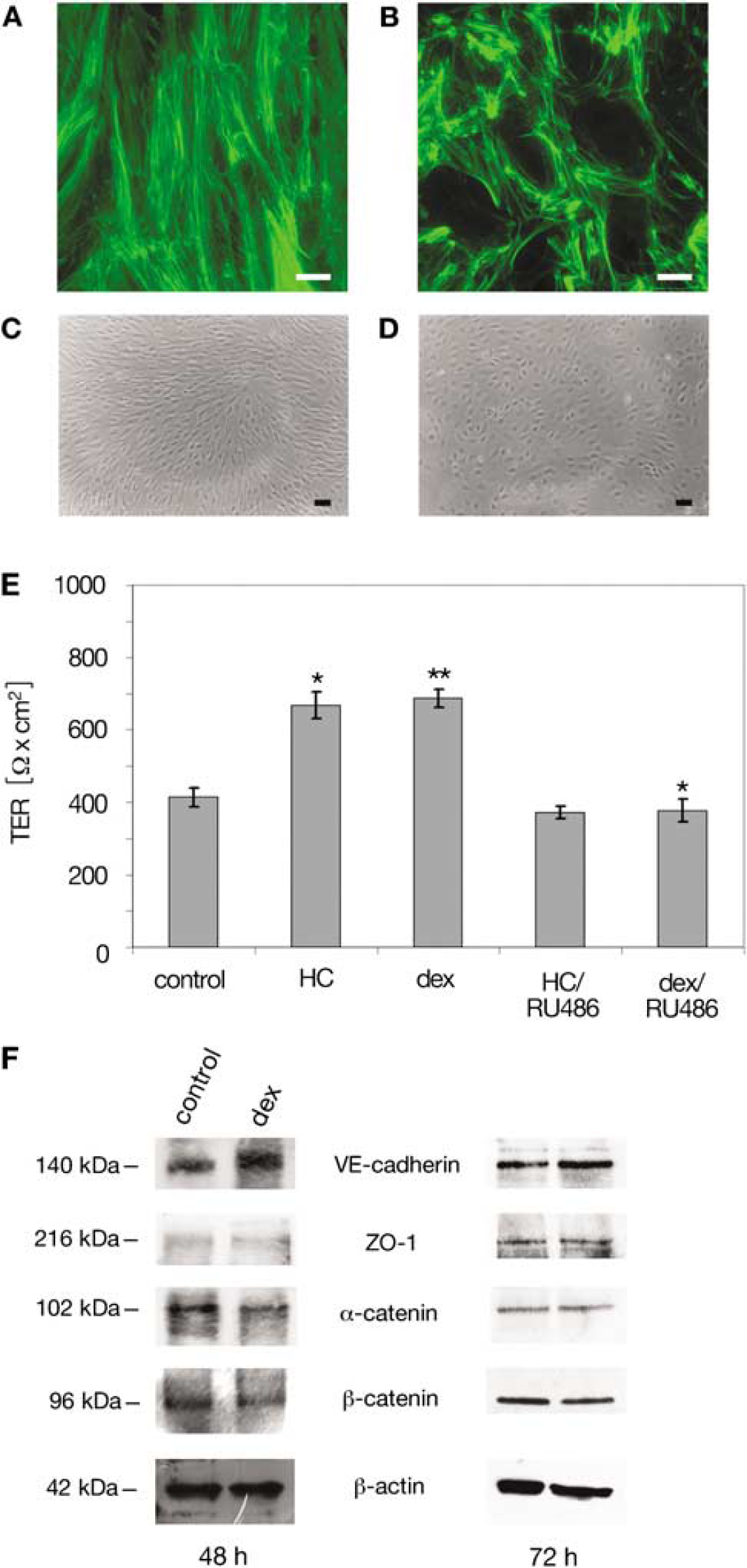
Effects of dexamethasone stimulation on cellular morphology and barrier properties in cEND brain capillary ECs. (
Glucocorticoid Induction of Barrier Properties in the Brain Endothelial Cell Line cEND
The AJ protein VE-cadherin is known to be a key mediator of endothelial barrier sealing and maintenance. To assess functional changes in the brain endothelium induced by the GC treatment regime used, we assessed TER in response to GC treatment (Figure 1E). For this, we assessed TER of untreated cells and compared the values with cells treated with hydrocortisone or dexamethasone (Figure 1E). We were able to show that treatment with hydrocortisone or dexamethasone increased TER values from 413±25 Ω cm2 to 668±36 Ω cm2 (hydrocortisone) and 687±26 Ω cm2 (dexamethasone).
Simultaneous treatment with hydrocortisone or dexamethasone and the GR antagonist RU486 did not lead to increased TER values as compared with untreated cells: 372±16 Ω cm2 (hydrocortisone) and 378±31 Ω cm2 (dexamethasone).
Dexamethasone Stimulates the Production of VE-cadherin Protein in cEND cells
We next examined alterations in the expression of cell—cell adhesion proteins, that is, VE-cadherin, the main AJ protein in ECs, the intracellular AJ plaque proteins α-catenin and β-catenin as well as ZO-1, the plaque protein shared by both AJ and TJ (Schnittler, 1998) in the presence or absence of dexamethasone for 48 and 72 h by immunoblotting.
Consistent with previous observations of occludin (Förster et al, 2005), we could observe a 149.1%±2.6% (48 h) and 130%±6% (72 h) increase in VE-cadherin protein levels in dexamethasone-treated cEND cells (Figure 1F), as determined by western blot and densitometric analysis, suggesting a positive effect of GCs on intercellular adhesive potential in the BBB model system. As described previously (Förster et al, 2007), positive effects on the other proteins investigated, α-catenin, β-catenin, and ZO-1, could not be observed (Figure 1F).
Glucocorticoid Regulation of VE-cadherin Gene Expression
The effects of GCs such as dexamethasone are classified into (1) direct genomic effects (transcriptional target gene induction by binding of the GR to target gene DNA) or (2) indirect genomic effects through protein—protein interaction of GR with other sequence-specific transcription factors (Beato, 1989). Thirdly, non-transcriptional regulation by the GR through signalling cascades has been shown to become effective with comparably slow kinetics (Reichardt et al, 2006). The positive effects of dexamethasone treatment on VE-cadherin protein levels and increased TER in the cEND cell line thus prompted us to further investigate whether the VE-cadherin gene would represent a potential direct GR target gene involved in GC-mediated regulation of BBB permeability. For this, we at first analyzed transcriptional activation of the VE-cadherin gene by RT-PCR.
To initially determine the time point of enhancement of the VE-cadherin transcription, we analyzed mRNA levels from dexamethasone-treated cEND cells: mRNA was isolated 24 h after single treatment and 48 h after repeated treatment (second treatment after 24 h). The induction of VE-cadherin mRNA reached its peak of 1.86±0.41-fold expression in dexamethasone-treated cEND cells 48 h after repeated treatment (Figure 2A). Earlier, at 24 h of single treatment, a 1.22±0.07-fold increase in transcript was detected (Figure 2A).
To scrutinize a direct GR-mediated transcriptional effect, based on binding of GR to the DNA, we tested the transcriptional activation of the VE-cadherin gene in the presence of dexamethasone or a combination of dexamethasone and the GR antagonist RU486, which interferes with the binding of the steroid—receptor complex to the major groove in the DNA (Agarwai, 1996). A 1.8±0.12-fold upregulation of VE-cadherin mRNA in confluent cEND cells was detected as a late GC effect after 48 h of dexamethasone (100 nmol/L) treatment by densitometric band evaluation (Figure 2B). This effect was not detectable in the presence of the GR antagonist RU486 (Agarwai, 1996) (Figure 2B). In contrast, a 24 h treatment did not show significant effects of either treatment, dexamethasone (0.96±0.041-fold expression) or dexamethasone/RU486 (1.01±0.03-fold expression), as compared with control cells, ruling out direct GR-mediated transcriptional regulation of VE-cadherin gene expression but rather pointing to indirect GC effects.
Dexamethasone Effects on Transactivation of Transfected VE-cadherin Promoter Construct in HEK293 and cEND Cells
To unambiguously rule out direct GR-mediated effects on VE-cadherin gene expression as a mode of GC action, we next tested transactivation of the VE-cadherin gene promoter after 24 h of treatment. For this, the transactivating effects of GC were tested on an isolated VE-cadherin promoter reporter construct (Lelievre et al, 2000) in HEK293 model cells and in cEND brain microvascular cells: we knew from previous publications (Lelievre et al, 2000) that the VE-cadherin promoter construct is difficult to transfect. Thus, it was necessary to establish a suitable transfection technique in HEK293 cells, which provide a valuable transfection model, being transfected with a high efficiency, and express endogenous GR (Oakley et al, 1999). Induction of VE-cadherin expression by dexamethasone was assessed with physiologic (50 and 100 nmol/L) and supraphysiologic (1 μmol/L) concentrations of the natural GCs hydrocortisone and dexamethasone, as well as with 100 nmol/L of either GC in the presence of the GR antagonist RU486. Treatment of HEK293 cells with 100 nmol/L dexamethasone stimulated reporter gene expression up to 3.37±0.83-fold (Figure 3A). A similar enhancement of VE-cadherin gene expression could be observed by treating with 1 μmol/L dexamethasone (4.56±0.5-fold transactivation). Treatment of HEK293 cells with 100 nmol/L hydrocortisone stimulated reporter gene expression up to 3.11±0.49-fold, whereas treatment with 1 μmol/L hydrocortisone led to similar, 3.64±0.6-fold, stimulation of reporter gene expression 24 h after GC administration (Figure 3A). Lower concentrations of glucocorticoids (50 nmol/L) led to a moderate transactivation in the case of both GCs, hydrocortisone (2.11±0.05-fold) and dexamethasone (1.77±0.22-fold), 24 h after GC administration. Upregulation by dexamethasone was nearly completely abolished in the presence of the GR antagonist RU486 in the case of both GCs, hydrocortisone (0.8±0.16-fold) and dexamethasone (0.9±0.1-fold), in the model cell line HEK293.
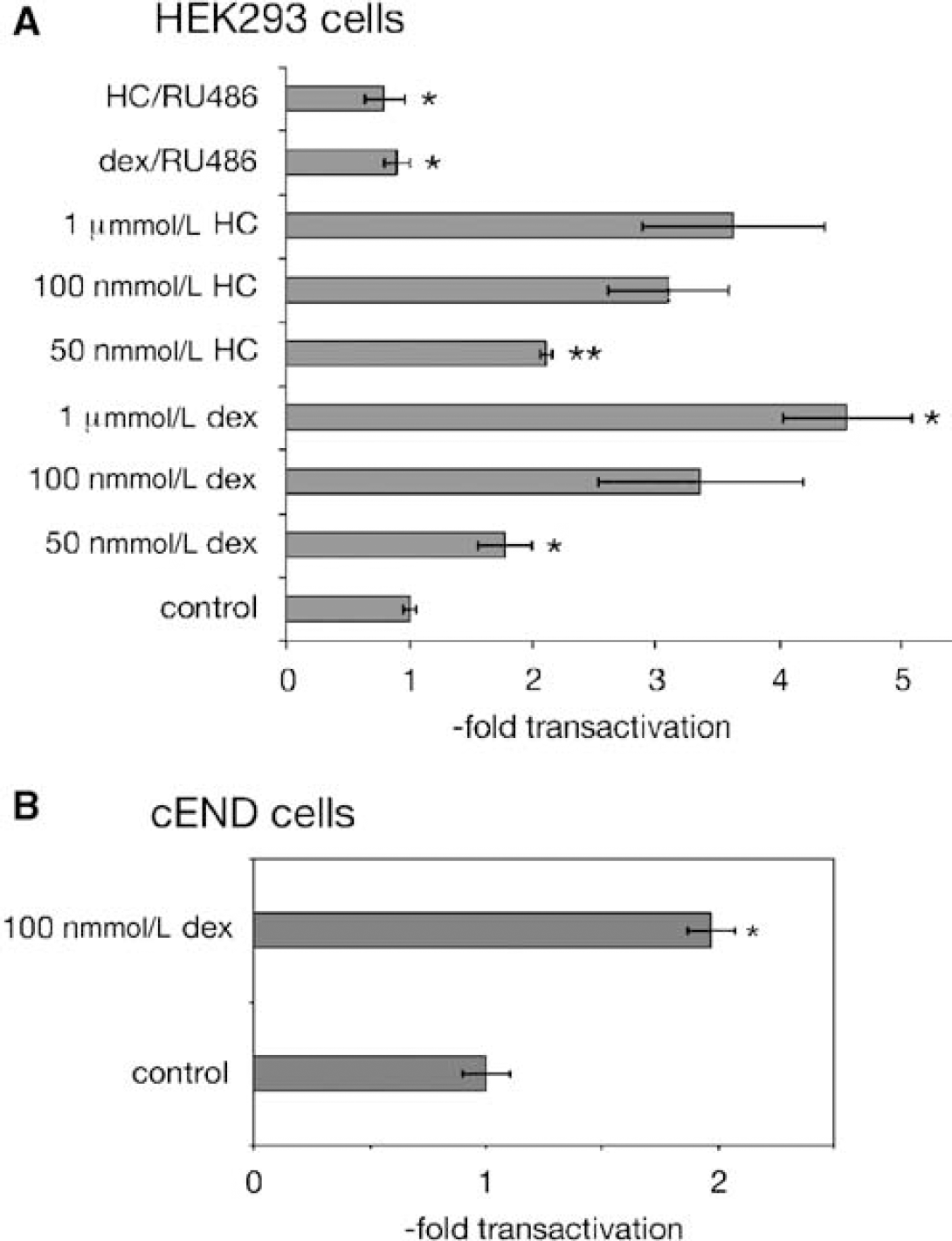
Transactivation of VE-cadherin by dexamethasone in the model system HEK293 cells and in cEND cells. (
The GC responsivity of the VE-cadherin promoter was subsequently tested in the EC context in confluent cEND cells, which were transfected with the −2486LUC promoter-reporter construct and treated with 100 nmol/L dexamethasone for 24 h. In transfected cEND cells, we could not repeat the gene transactivation detected in HEK293 model cells but observed only a 1.97±0.1-fold transactivation after 24 h of treatment with 100 nmol/L dexamethasone (Figure 3B). As transactivation rates in endothelial cEND cells were not comparably high in HEK293 model cells and gene induction was not detected before 24 h of incubation with GC, the data obtained from promoter-reporter gene assays equally pointed to indirect GC effects on VE-cadherin expression rather than a direct transcriptional regulation of the VE-cadherin gene by GR.
Glucocorticoid Treatment Increases VE-cadherin Anchorage to the Actin Cytoskeleton
The endothelial barrier function is largely dependent on EC junctions and the maturation of AJs requires an anchorage of intercellular VE-cadherin molecules to the actin cytoskeleton by means of the cytoplasmic region through catenins (Gumbiner, 1996). It is possible to detect the amount of VE-cadherin anchored to the cytoskeleton in the detergent-insoluble cell fraction through the VE-cadherin cytoskeletal association assay, resulting in a fractionation of the cell protein compounds by the Triton X-100 detergent. Triton X-100 dissolves the cell membrane so the detergent-soluble fraction contains all cytosolic proteins. The Triton X-100 insoluble fractions were used for immunoblotting to examine the levels of VE-cadherin protein associated with the cytoskeleton in the presence or absence of dexamethasone. As a result of the cell fractionation, we found an increase of 42%±24.1% in the cytoskeletal-associated VE-cadherin in dexamethasone-treated cells as compared with control cells (Figures 4A and 4B). As a control for equal loading, β-actin western blot was performed (Figure 4A).
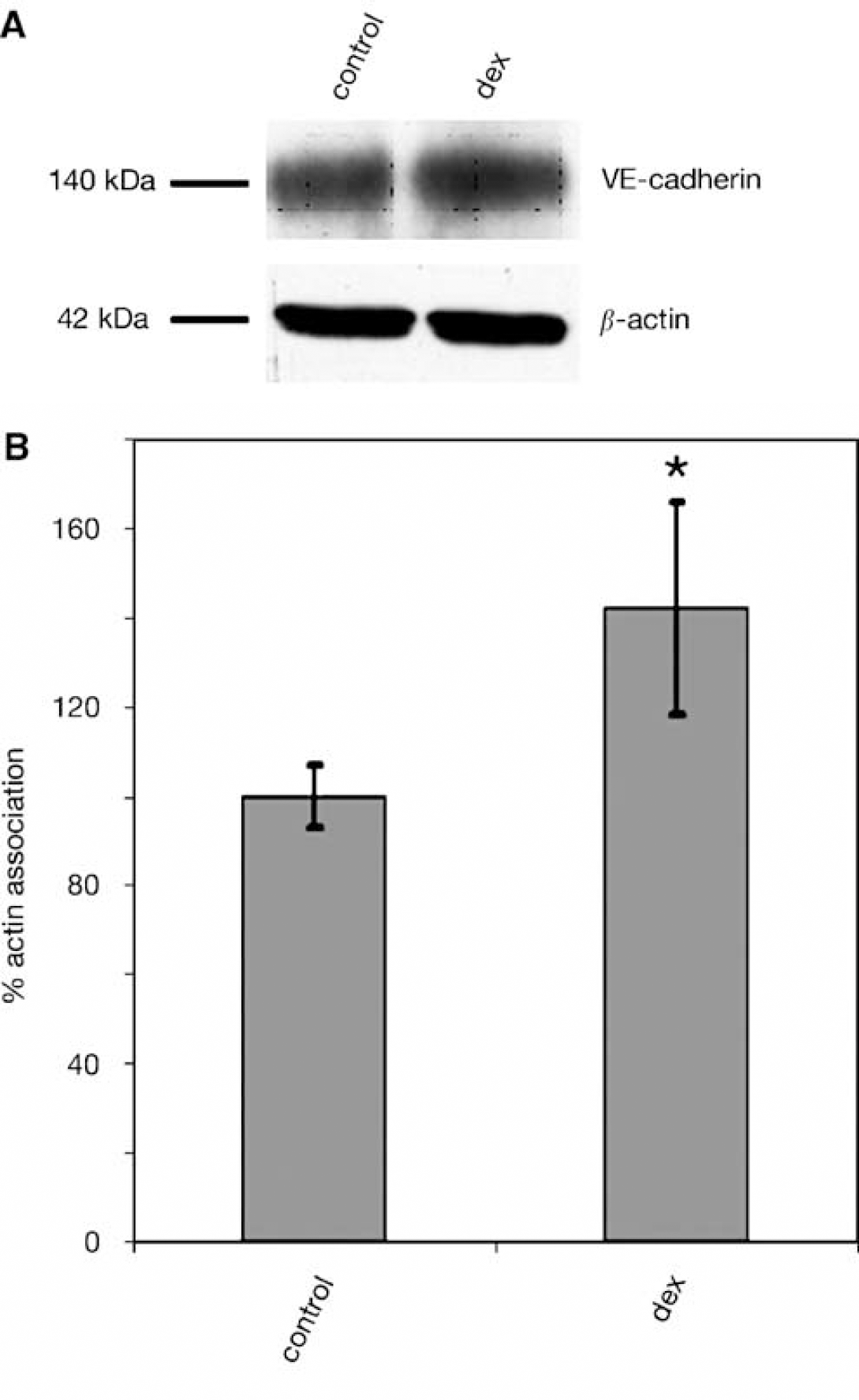
GC treatment results in reinforcement of cytoskeletal VE-cadherin anchorage. (
Discussion
The integrity of the BBB is compromised in many disorders of the human central nervous system. Blood—brain barrier breakdown under conditions of neuroinflammation and cerebral ischemia, but also traumas and brain tumors, leads to loss of the protective function of the barrier. In BBB breakdown, one of the first observable changes is the loss of intercellular adhesion and concomitant increase of permeability (Hawkins and Davis, 2005). Furthermore, disruption of cell adhesion is reported in pathologic conditions such as meningitis (Sellner and Leib, 2006) and cerebral malaria (Pino et al, 2005). These studies show that the precise regulation of cell—cell interactions is a crucial aspect of maintaining the normal structure and function of the BBB.
Although therapeutic strategies for diseases with impaired BBB function include treatment with GCs (Engelhardt, 2000), the mechanism explaining GC action is still unclear. Although most publications focus on the action of GCs in inflammation, the mechanism of the observed barrier-tightening effects of GC (Burnham et al, 1991; Förster et al, 2005; Hoheisel et al, 1998; Romero et al, 2003) remain elusive. In our previous works, we showed a new aspect in GC action, the upregulation of the TJ protein occludin by GCs by direct transcriptional gene activation by means of the GR (Förster et al, 2005, 2006). In this study, we tested whether further components of the apical junctional complex might represent potential GR target genes in the immortalized brain EC line cEND. Positive GC effects on the AJ transmembrane protein VE-cadherin were detected and prompted us to further investigate the molecular basis of GC action on VE-cadherin gene induction and anchorage of VE-cadherin protein to the cytoskeleton.
VE-cadherin protein levels were significantly increased as a late effect of GC administration. We could not detect any changes in VE-cadherin expression or intracellular localization as an early response to GC. By promoter-transactivation studies in vitro, in a complementary manner, we could not document substantial transactivation of a reporter gene under the control of the VE-cadherin promoter as an early response to physiologic GC concentrations. For this, gene transactivation was monitored in the model cell line HEK293 as well as in cEND brain microvascular cells. Minor effects on VE-cadherin gene induction were observed only as a late steroid response (>24 h) so that the data obtained rather point to indirect GC effects on VE-cadherin expression than a direct transcriptional regulation of the VE-cadherin gene by GR.
The TJ, AJ, and the cytoskeleton form an integrated functional unit through multiprotein complexes (Gumbiner, 1996). For example, the cadherin proteins of the AJ are structurally connected to F-actin through β-catenin and α-catenin (Gumbiner, 1996). We thus wanted to prove whether the observed reinforcement of barrier functions could be correlated to a more firm anchorage of the VE-cadherin-based AJ to the actin cytoskeleton. To answer this question, we examined the VE-cadherin association with the cytoskeletal fraction by detergent fractionation. By this experiment, we were able to determine the level of VE-cadherin protein anchored to the cytoskeleton in detergent-insoluble fractions of cell lysates from dexamethasone-treated and untreated cEND cells. It was possible to show that a late cellular response to GC treatment consisted of increased levels of VE-cadherin protein concomitant with an increased relative recruitment of VE-cadherin from the cytosol to the cytoskeleton-associated cell fraction. In a complementary manner, dexamethasone administration caused a reorganization of F-actin into a cortical ring in the presence of the steroid. Similar changes in cytoskeletal and tight junctional proteins have been described to correlate with decreased permeability in dexamethasone-treated brain ECs from rat (Romero et al, 2003) and porcine origin (Torok et al, 2003) in vitro in the past. For the cell line cEND, we had documented previously (Förster et al, 2005) that the GC hydrocortisone induces morphologic changes from a spindle-shaped morphology to a cobblestone morphology and stimulates the monolayer TER, which is a reliable in vitro measurement of TJ sealing. By this morphologic change, the calculated junctional length available for paracellular diffusion across the monolayer was determined to have shortened by approximately 50% as compared with untreated cells (Förster et al, 2005), and this decrease in junctional length was assumed to be chiefly responsible for permeability reduction. In this study, we show an increase in VE-cadherin expression and an increased anchorage of VE-cadherin protein to the actin cytoskeleton concomitant with the formation of a cortical actin ring as a late cellular response to GCs and resulting reinforcement of barrier function. Interestingly, although not in the brain vascular endothelial system, studies on intestinal absorptive cells led by Madara et al gave first evidence that the barrier function of TJs was correlated with the degree of junctional protein association with actin filaments (Madara et al, 1986). They showed that an altered distribution of actin coincided with perturbed paracellular permeability, which was evidenced by decreased transepithelial resistance. Several studies have also shown the importance of the junction-associated actin ring for endothelial barrier formation, indicating that depolymerization of F-actin and fragmentation of the endothelial cortical actin ring lead to increased permeability of the endothelial barrier (Schnittler et al, 1990; Waschke et al, 2004). We now complement this observation with the finding that improvement of endothelial barrier function by GC is accompanied by reinforcement of the AJ and the VE-cadherin-actin connection: our study shows that the administration of the GCs dexamethasone and hydrocortisone strengthens barrier properties in the brain EC line cEND. This can be partially accounted for by reinforcement of VE-cadherin anchorage to the actin cytoskeleton and changes in cellular morphology, resulting in a more cobblestone cellular morphology as observed in tight epithelia or primary ECs of the BBB (Lossinsky and Shivers, 2004). Additionally, we previously showed that GCs directly induce the formation of the endothelial TJ protein occludin (Förster et al, 2005, 2006), thus providing a pro-barrier effect.
Taken together, our results indicate that the GC upregulation of VE-cadherin is an indirect steroid response that is associated with the formation of a cortical actin ring leading to a tight barrier. The adhesive interactions between VE-cadherin molecules might be primary, whereas interactions between apposing occludin and claudins may become secondary or supplementary components in maintaining a functional TJ complex and these remain to be elucidated by further studies. As cellular GC effects were however detected as a late response to GC treatment (48 to 72 h of incubation), the GC treatment could induce or regulate the expression of a whole set of structural and/or regulatory factors involved in cellular differentiation and redistribution of actin from stress fibers to a peripheral band resulting in reinforcement of endothelial barrier properties. For instance, changes in the activity of Rho family GTPases with a possible increase in Rac1, associated with an increase in cortical actin (Adamson et al, 2002), could be envisaged. Further characterization of GC targets upstream of VE-cadherin may provide important insights into the hormonal control of the endothelial junctional complex.
Based on our work, we provide strong evidence that remodelling of the EC adhesion complex might be a general feature underlying the hormone-mediated improvement of BBB qualities and that the cell biologic steps described are required for the steroid-induced remodelling of the apical junction architecture that leads to functional TJs. Whether endogenous GCs contribute to BBB maturation in development by the effects observed remains to be investigated in the future.
Footnotes
Acknowledgements
The VE-cadherin luciferase reporter vector −2486LUC was a generous gift of Dr Fabrice Soncin (CEA-Grenoble, Grenoble, France). The authors are grateful to Eva-Maria Klute for excellent technical assistance.