
Abstract
Present knowledge about hemodynamic and metabolic changes after subarachnoid hemorrhage (SAH) originates from neuromonitoring usually starting with aneurysm surgery and animal studies that have been focusing on the first 1 to 3 h after SAH. Most patients, however, are referred to treatment several hours after the insult. We examined the course of hemodynamic parameters, cerebral blood flow, and tissue oxygenation (ptiO2) in the first 6 h after experimental SAH. Sixteen Sprague–Dawley rats were subjected to SAH using the endovascular filament model or served as controls (n = 8). Bilateral local cortical blood flow, intracranial pressure, cerebral perfusion pressure, and ptiO2 were followed for 6 h after SAH. After induction of SAH, local cortical blood flow rapidly declined to 22% of baseline and returned to 80% after 6 h. The decline of local cortical blood flow markedly exceeded the decline of cerebral perfusion pressure. ptiO2 declined to 57%, recovered after 2 h, and reached ≥140% of baseline after 6 h. Acute vasoconstriction after SAH is indicated by the marked discrepancy of cerebral perfusion pressure and local cortical blood flow. The excess tissue oxygenation several hours after SAH suggests disturbed oxygen utilization and cerebral metabolic depression. Aside from the sudden increase of intracranial pressure at the time of hemorrhage and delayed cerebral vasospasm, the occurrence of acute vasoconstriction and disturbed oxygen utilization may be additional factors contributing to secondary brain damage after SAH.
Introduction
Cerebral arteries have been observed to react to subarachnoid hemorrhage (SAH) in a biphasic pattern, acute vasoconstriction starting immediately after SAH, delayed vasospasm several days after the insult (Brawley et al, 1968; Bederson et al, 1998). In the past decades, clinical and experimental research has focused on delayed vasospasm, as it was believed that poor outcome after SAH was mainly determined by vasospasm-induced ischemia. Until now, however, no effective treatment for delayed vasospasm or spasm-induced infarction has been brought forth. In a recent pilot trial, the endothelin-antagonist clazosentan attenuated or even reversed cerebral vasospasm but did not significantly inhibit delayed ischemic infarction or improve outcome (Vajkoczy et al, 2005). Similarly, tirilazad, an inhibitor of lipid peroxidation significantly decreased symptomatic vasospasm but did not decrease the incidence of cerebral infarction or significantly improve clinical outcome (Jang et al, 2008). These findings show that the clinical course and outcome after aneurysmal SAH are not solely determined by delayed cerebral vasospasm.
Apart from the reduction of cerebral blood flow (CBF), a depression of cerebral oxygen metabolism (CMRO2) has been observed in patients after aneurysmal SAH (Grubb et al, 1977; Hayashi et al, 2000). Although the extent of this phenomenon positively correlated with the patients' clinical state and the amount of subarachnoid blood, it has been observed in patients of all clinical grades and with more or less subarachnoid blood (Jakobsen et al, 1991; Carpenter et al, 1991). Whether there is a relationship between cerebral vasospasm and the depression of oxygen metabolism is discussed with controversy. Voldby et al found a concomitant decrease of CMRO2 with arterial caliber, indicating secondary metabolic impairment due to cerebral vasospasm (Voldby et al, 1985). Other authors did not observe an additional depression of CMRO2 with the appearance of delayed vasospasm and advocated a primary reduction of metabolism by SAH (Grubb et al, 1977; Carpenter et al, 1991; Frykholm et al, 2004). All of these studies, however, measured CBF and metabolic parameters in patients at various times after SAH and after hospital admission. Rasmussen et al obtained preoperative measurements of CBF and CMRO2 before aneurysm surgery, including individuals up to 24 h after SAH (Rasmussen et al, 1999). Other authors included patients up to several days after SAH (Frykholm et al, 2004; Jakobsen et al, 1991). These studies did not cover the initial posthemorrhagic period.
Information about the acute stage after SAH has to be drawn out of animal studies. Concerning the examination of metabolic alterations after experimental SAH emphasis, again, was put on changes during the period of late arterial vasospasm (Sahlin et al, 1987; Solomon et al, 1987). Some studies, however, concentrated on the insult itself and the period immediately thereafter. Most of those studies—using a variety of experimental models—examined the course of hemodynamic parameters and CBF (Bederson et al, 1998; Schwartz et al, 2000) and metabolic changes (Prunell et al, 2003) in the first 60 to 90 mins after SAH. Jackowski et al monitored CBF and cerebral perfusion pressure (CPP) for 3 h after SAH (Jackowski et al, 1990).
In clinical practice, SAH is rarely diagnosed within the first 3 h after the insult. In the majority of high-grade SAH patients, diagnosis is established after 3 to 6 h, sometimes even later (Audebert et al, 2005). Although of high clinical relevance, there is a lack of information regarding this period after SAH.
The present study was designed to gather information about pathophysiological sequences and processes in this particular period. The endovascular filament model in rats was used, because this experimental model has been found to be particularly suitable for the examination of the acute stage of SAH (Gules et al, 2002; Schwartz et al, 2000; Sehba et al, 1999; Bederson et al, 1998). It allows continuous monitoring of hemodynamic parameters and cerebral pathophysiological changes without violation of the intracranial vault and without repositioning the animal or monitoring probes. We extended the observation time to 6 h after induction of SAH, as it was the aim of this study to characterize pathophysiological sequences in this clinically relevant period, particularly focusing on cerebral perfusion and brain tissue oxygenation as a parameter of CMRO2.
Materials and methods
For the experiments, 16 male Sprague–Dawley rats (250 to 300 g body weight), purchased from Harlan Winkelmann (Borchen, Germany) were used. All experiments were approved by the regional authorities and the district government of Bavaria, Germany.
Animal Preparation and Monitoring
The animals were anesthetized with 4% isoflurane, orally intubated and mechanically ventilated with an air–oxygen mixture to maintain normal arterial blood gases. After induction of anesthesia, isoflurane was reduced to 2.5% for surgical procedures and to 1.5% from 30 mins before SAH until the end of the monitoring period. Temporalis muscle and rectal probes were used to monitor temperature throughout the experiment. A thermostatically regulated, feedback-controlled heating lamp was used to maintain temporalis muscle and rectal temperature at 37°C. The tail artery was cannulated for continuous measurement of mean arterial blood pressure and for blood sampling. Arterial blood gases were measured 30 and 5 mins before and in hourly intervals after induction of SAH.
Laser Doppler Flowmetry, Intracranial Pressure, and Tissue Oxygenation
A two-channel laser-Doppler flowmeter (LDF) (MBF3D; Moor Instruments, Axminster, England) was used for continuous bilateral monitoring of local cortical blood flow (LCBF) in the area of the cerebral cortex supplied by the MCA. To place the LDF probes, burr holes were drilled 5mm lateral and 2mm posterior to the bregma without injury to the dura mater.
For measurement of brain tissue oxygenation (ptiO2), a burr hole was drilled over the right frontal cortex 3mm lateral and 1mm anterior to the bregma. The dura was opened without damage to the cerebral cortex. A Licox small animal brain tissue oxygenation probe (Integra Neurosciences, Plainsboro, NJ, USA) was inserted 3mm into the brain and fixed with dental cement. Calibrations were made according to the manufacturer's recommendations. For measurement of intracranial pressure (ICP), a further burr hole was drilled over the left frontal cortex 3mm lateral and 0.5mm anterior to the bregma.
After all burr holes were completed, the animals were placed in a supine position with the head fixed in a stereotactic frame with earbars. A rectangularly bent laser-Doppler probe was positioned in each burr hole with a micromanipulator. An intraparenchymal Camino ICP probe (Integra Neurosciences, Plainsboro, NJ, USA) was advanced 2mm into the brain by a third micromanipulator.
Induction of Subarachnoid Hemorrhage
Subarachnoid hemorrhage was induced by the endovascular puncture method (Veelken et al, 1995; Bederson et al, 1995). After surgical exposure of the right cervical carotid bifurcation, temporary aneurysm clips were placed on the common and internal carotid artery (ICA). A 3–0 Prolene filament (Ethicon, Inc., Somerville, NJ, USA) was inserted into the external carotid artery and fixed with a silk ligature and the temporary clips were removed. After a stabilization period of 30 mins, the filament was advanced into the ICA until ipsilateral LCBF decreased indicating that the tip of the filament was at the intracranial division of the ICA occluding the origin of the MCA. The filament was then pushed 2 to 3mm further for intracranial vessel perforation in the ICA–ACA region. The suture was then quickly withdrawn into the external carotid artery to allow reperfusion and development of SAH. Subarachnoid hemorrhage was indicated by a rapid bilateral decrease of LCBF and increase of ICP.
Experimental Groups
The rats were randomly assigned to one of two groups (n = 8 for each group): (1) SAH by a 3–0 Prolene filament and (2) sham-operated controls. In the latter group, a 3–0 filament was inserted into the external carotid artery and advanced via the ICA until LDF showed an ipsilateral decline but was not advanced further.
Termination of the Experiment and Wound Closure
Six hours after induction of SAH, monitoring was stopped, the ICP probe, laser-Doppler probes, and arterial catheter were removed, and the wounds were closed with a skin suture. Isoflurane was withdrawn, and the animals were allowed to wake up.
Quantification of Subarachnoid Blood and Cerebral Infarction
After 24 h, the animals were again anesthetized with isoflurane followed by an intraperitoneal injection of 50mg sodium thiopental. The animals were transcardially perfused with 4% paraformaldehyde. The brains were removed, and the amount of subarachnoid blood was determined using a semiquantitative scale (SAH score) as follows: (0) no blood visible; (1) traces of blood visible, no blood clot; (2) unilateral clot; (3) generalized bilateral basal blood clot; (4) intracerebral hematoma with or without subarachnoid blood.
Thereafter, the brains were embedded in paraffin and cut into 4-µm-thick coronal sections at 400-µm intervals, and the brain slices were stained with cresyl violet. The slices were microscopically scanned for early signs of cerebral infarctions, lacunar or territorial, and for intracerebral hemorrhage. Three defined parts of the CA1 region of the hippocampus (bregma −3.24, −4.92, −6.12) (Paxinos and Watson, 2005) were bilaterally analyzed for surviving neurons, and their number per field (0.2 × 0.3mm) was counted. Neurons located completely inside the window and neurons that crossed only through its top or right side were counted. Cells crossing the bottom and left side of the frame were not counted. Viable (counted) cells fulfilled the following criteria: sharply delineated nucleus, clearly distinguishable nucleolus located centrally within the nucleus, and less than one third of the neuron surrounded by confluent vacuolization (Stummer et al, 1994).
Correlations
Histopathological damage and final values of ptiO2 (360 mins after SAH) were compared with CPP and LCBF for each time point (1 to 360 mins after SAH) using a Pearson correlation.
Statistical Analysis
Statistical analysis was performed with SPSS 14.0 (SPSS, Inc., Chicago, IL, USA). Physiologic data for each time point, LCBF, ICP, and ptiO2 data were analyzed by a Student's t-test. Correlations were calculated using a Pearson correlation. A P>0.05 was considered to be statistically significant. The results are presented as mean±s.d.
Results
Physiologic Parameters
There were no significant differences between the groups regarding pH, pCO2, and pO2. Values at the end of surgical preparation, immediately before SAH, 1 and 3 h after SAH are depicted in Table 1.
Arterial blood gases were first measured after completion of surgical procedures and mechanical ventilation was adjusted
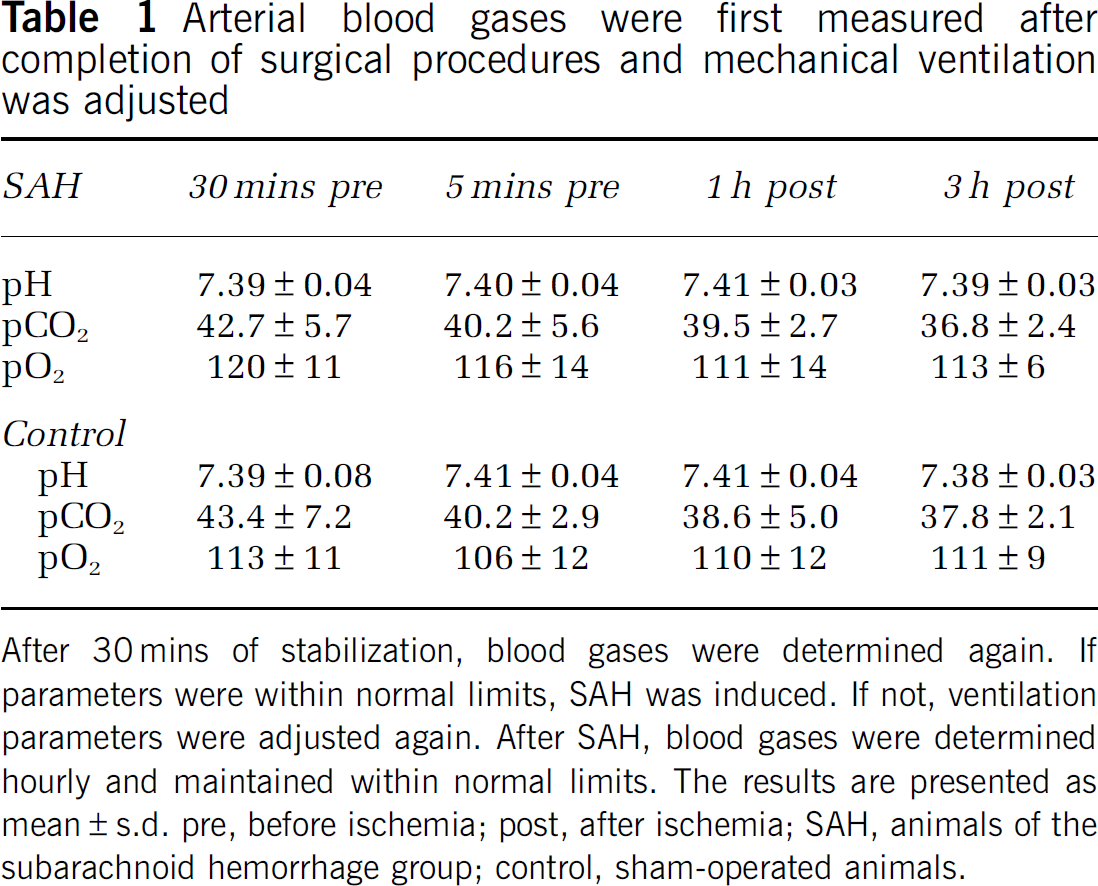
After 30 mins of stabilization, blood gases were determined again. If parameters were within normal limits, SAH was induced. If not, ventilation parameters were adjusted again. After SAH, blood gases were determined hourly and maintained within normal limits. The results are presented as mean±s.d. pre, before ischemia; post, after ischemia; SAH, animals of the subarachnoid hemorrhage group; control, sham-operated animals.
Intracranial Pressure, Mean Arterial Blood Pressure, Cerebral Perfusion Pressure
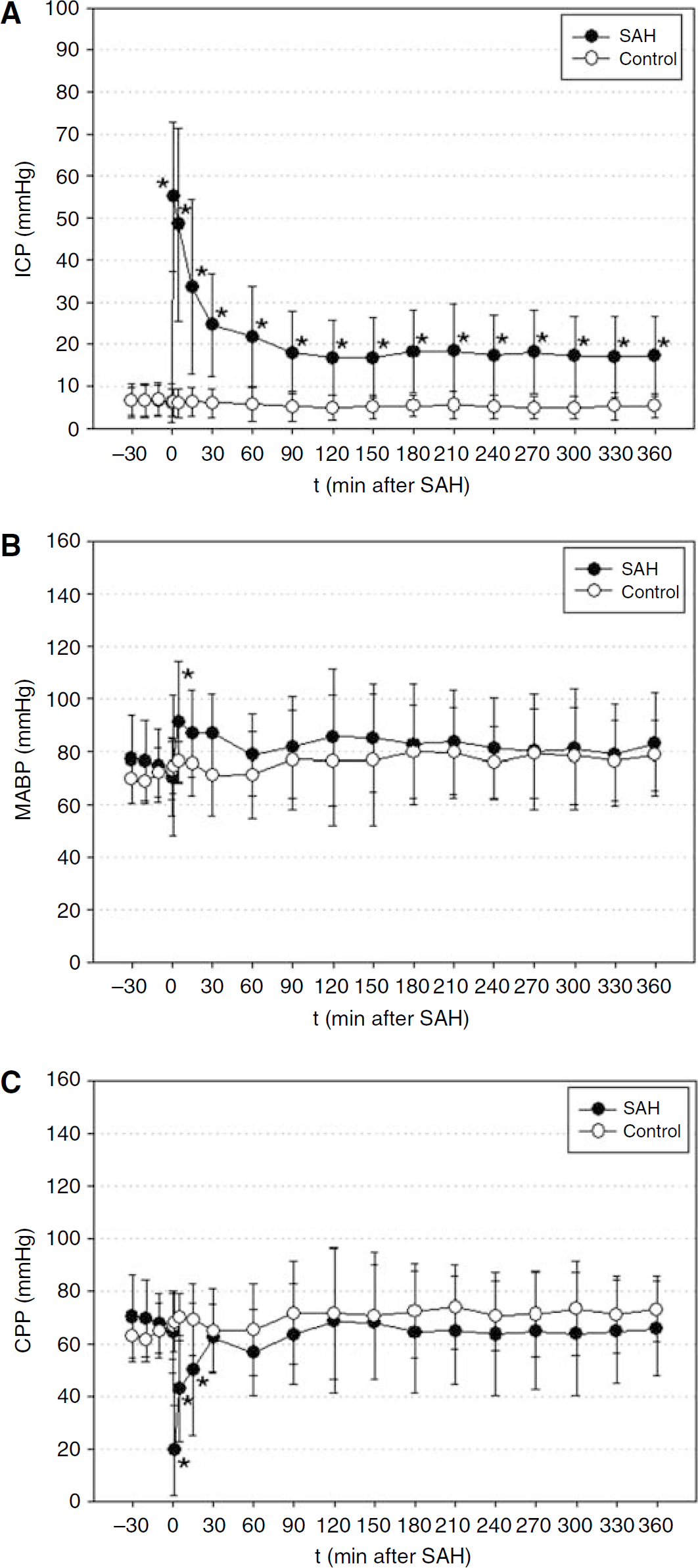
Intracranial pressure rapidly increased in the SAH group from a baseline of 6±5mmHg to a maximum of 55±18mmHg 1 min after induction of SAH and returned to 22±12mmHg after 1 h and 17±10mmHg after 6 h. In the control group, baseline ICP was 6±3mmHg and did not show relevant changes throughout the monitoring time. The increase of ICP in the SAH group was significant compared with the control group from induction of SAH until the end of the observation time (Figure 1A).
(
Mean arterial blood pressure increased in the SAH group from a baseline of 70±14mmHg to a maximum of 91±23mmHg 5 mins after SAH and gradually returned to 82±19mmHg at the end of the observation period. Baseline mean arterial blood pressure in the control group was 73±11mmHg and reached 79±15mmHg after 6 h. The increase of mean arterial blood pressure was significant 5 mins after SAH (Figure 1B).
Cerebral perfusion pressure declined in the SAH group from a baseline value of 64±16mmHg to a minimum of 19±17mmHg 1 min after SAH and recovered to 57±16mmHg after 1 h and 66±18mmHg after 6 h. Baseline CPP in the control group was 66±7mmHg and increased slightly to reach an endpoint of 73±14mmHg after 6 h. The decline of CPP in the SAH group was significant compared with the control group from the onset of SAH until 15 mins thereafter (Figure 1C).
Local Cortical Blood Flow
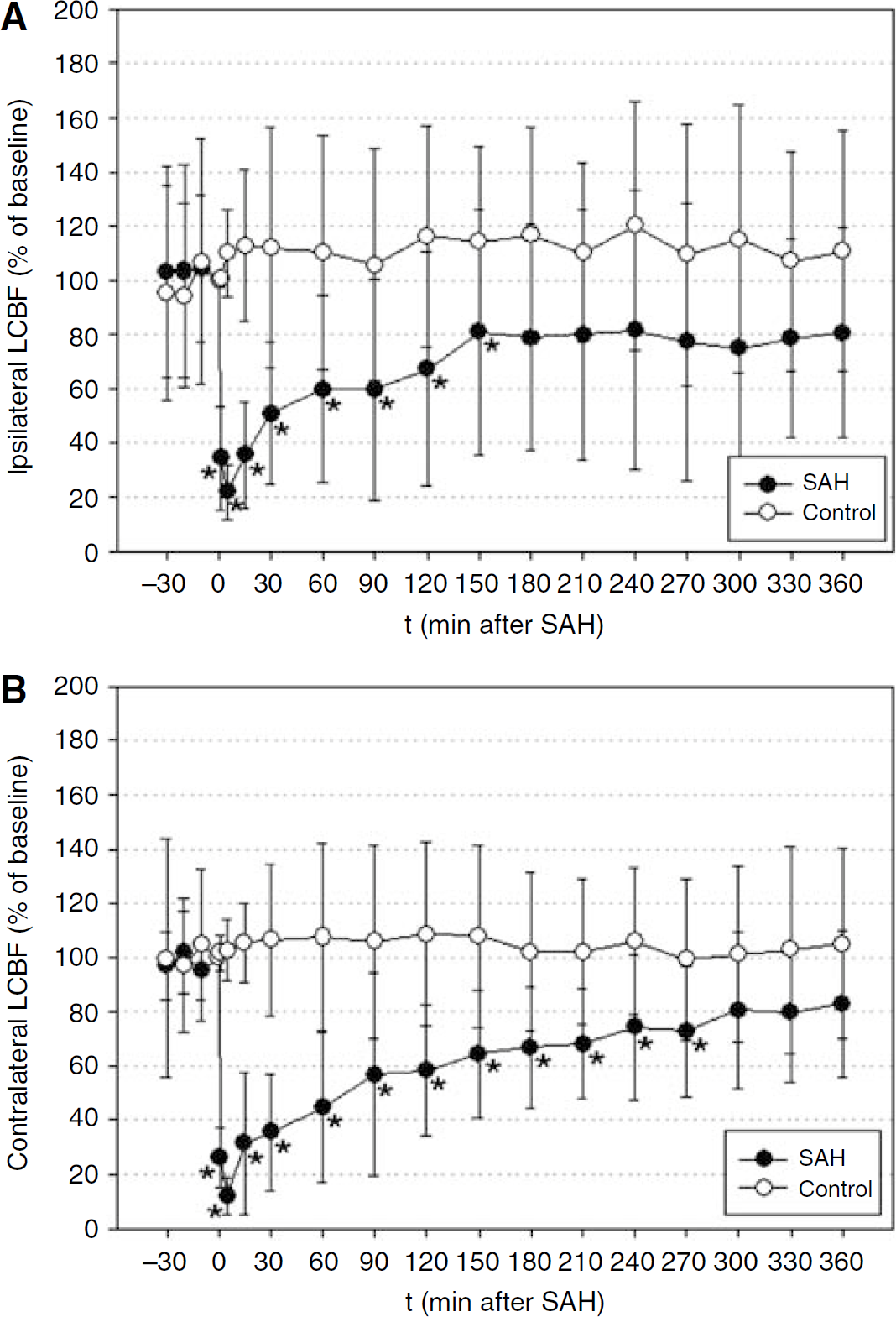
Ipsilateral LCBF declined to a minimum of 22%±10% of baseline 1 min after SAH, recovered to 60%±34% after 60 mins, and increased further to 81%±38% at the end of the observation period. In the control group, LCBF slightly increased to 110%±43% after 60 mins and stayed at this level until the end of the observation period. The decrease of LCBF was significant compared with the control group from the onset of SAH until 150 mins thereafter (Figure 2A).
(
Contralateral LCBF declined to a minimum of 12%±6% of baseline 5mins after SAH and recovered to 45%±28% after 60 mins and increased further to 83%±27% at the end of the observation period. In the control group, contralateral LCBF also slightly increased to 105%±35% after 60mins and remained at this level to reach 105%±34% at the end of the observation period. The decrease of LCBF was significant compared with the control group from the onset of SAH until 270mins thereafter (Figure 2B).
Brain Tissue Oxygenation
Brain tissue oxygenation (ptiO2) declined to a minimum of 56%±24% of baseline and recovered to 74%±34% after 60 mins, followed by a further continuous increase to 99%±37% of baseline 120mins after SAH and 143%±45% and 144%±44%, 330 and 360mins after SAH, respectively. In the control group, ptiO2 slightly increased to 103%±30% of baseline at the end of the observation period. The differences between the groups were significant 5 and 15mins after SAH and 330 and 360mins after SAH, respectively (Figure 3).
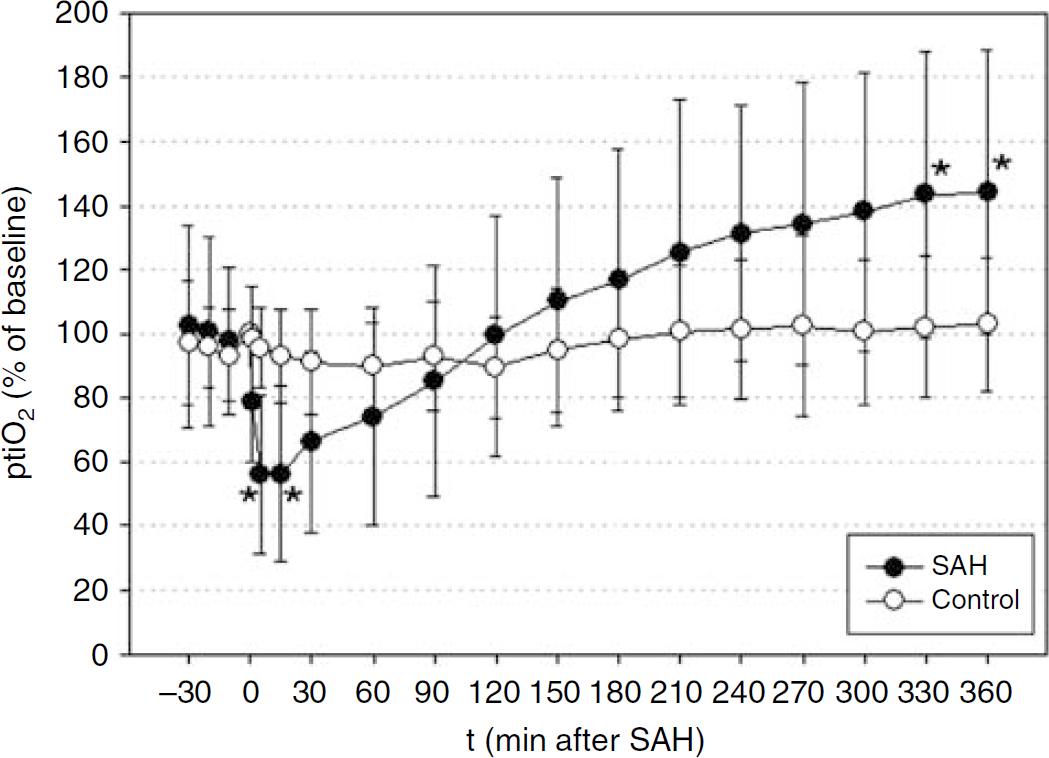
In contrast to the course of cerebral blood flow, brain tissue oxygenation (ptiO2) had recovered fully after 2 h and continued to increase to a plateau of over 140% of baseline at the end of the observation period. Values are presented as mean±s.d. (empty circles represent control group and filled circles represent subarachnoid hemorrhage (SAH) group; *P>0.05 versus control group for each time point).
Subarachnoid Blood and Histopathological Damage
In both groups, no animal died during the acute stage of the experiment or within 24h after SAH after termination of anesthesia. The brains of all SAH animals had significant amounts of blood in the basal subarachnoid space. In six animals, unilateral blood clots were found (score 2), and in two animals, the entire basal subarachnoid space was filled with blood (score 3). No blood was visible in the subarachnoid space over the convexities. The brains of control animals showed no traces of blood in the subarachnoid space. Microscopic examination of perfusion fixated brain slices showed the effects of the positioning of the ICP probe in the frontal cortex but no other intracerebral lesions. In particular, we did not find any signs of territorial or lacunar infarction or intracerebral hemorrhage. In the control group, 62±8 viable neurons per visual field were counted in the right hippocampal CA1 field and 61±8 neurons in the left CA1 field. In SAH animals, 49±8 neurons were counted on the right side (–21%) and 47±6 neurons on the left side (–23%) (P<0.01).
Correlations
There was no significant correlation between hippocampal damage and LCBF or CPP, nor between hippocampal damage and the final ptiO2 value 360 mins after SAH. ptiO2 360 mins after SAH was negatively correlated with CPP values in the first minutes after SAH. This correlation was significant for the CPP value 15 mins after SAH (r =–0.86; P > 0.01). ptiO2 and LCBF values of both hemispheres were negatively, but not significantly, correlated in the first 15 mins after SAH. Thereafter, the correlation coefficients were positive for LCBF values of both hemispheres and significant for ipsilateral values from 150 to 360 mins after SAH.
Discussion
The present experiments were conducted to gather further information about pathophysiological changes in the first hours after SAH, with particular focus on the period when the majority of SAH patients are admitted to treating units. In patients with severe SAH, this usually takes place between 3 and 6 h after the insult (Audebert et al, 2005). The data were obtained by continuous monitoring of ICP, brain tissue oxygenation, bilateral LCBF, and hemodynamic parameters for 6 h after SAH.
Cerebral Blood Flow and Acute Vasoconstriction
Induction of SAH resulted in an immediate and steep decrease of CPP. However, LCBF decreased even further and, while CPP recovered fast and almost completely, the decline of LCBF recovered slowly and exceeded the decline of CPP throughout the entire observation period. The rapid bilateral reduction of LCBF in the first seconds after SAH is certainly caused by the simultaneous increase of ICP and decrease of CPP. However, the gradual and incomplete recovery of LCBF and the discrepancy of CPP and LCBF starting only a couple of minutes after SAH can be best explained by a generalized vasoconstriction.
The occurrence of a biphasic vascular reaction after SAH consisting of acute vasoconstriction and delayed vasospasm has been described previously by several authors (Brawley et al, 1968; Peerless et al, 1982; Delgado et al, 1985). Concerning the decrease and incomplete recovery of cerebral perfusion, our findings in the first 60 mins after SAH are in accordance with the results of Sehba et al and Bederson et al in their studies about the early phase of vasoconstriction after SAH (Sehba et al, 1999; Bederson et al, 1998).
For further discussion of laser-Doppler data, we assumed that (1) baseline laser-Doppler flow reflects the normal tissue perfusion of approximately 55 ml per 100 g per min, (2) changes of LDF reflect relative changes of LCBF linearly (Dirnagl et al, 1989), and (3) bilateral parallel changes of LDF in independent vascular territories indicate a global reduction of CBF. Schmid-Elsaesser et al methodologically examined the endovascular filament model of MCA occlusion in rats. By measuring LDF over both hemispheres, they elaborated that a unilateral decrease of flow after advancing the filament indicates MCA occlusion. The reason for an immediate bilateral change was vessel perforation causing SAH in all cases (Schmid-Elsaesser et al, 1998). In our experiments, we also observed a unilateral decrease of LDF and used it as an indicator for the tip of the filament being located at the division of the carotid artery occluding the MCA. Continuous LDF via probes positioned over both hemispheres supplies data about relative changes of blood flow in territories that are essentially independent of each other. Consequently, continuous LDF has been experimentally validated and has proven useful even for controlling predetermined values of CBF in animal models of global ischemia (Dirnagl et al, 1993). In the absence of profound cardiovascular reactions, bilateral and parallel reactions of LDF can, therefore, be considered to be a valid method for the measurement of relative changes of CBF.
If CBF drops below 22 ml per 100 g per min, the brain's functional activity ceases, but cellular integrity is maintained. If it declines below one third of normal, neurons quickly switch to an anaerobic metabolism, and the development of infarction depends on the duration of the perfusion deficit (Siesjo, 1992; Jones et al, 1981). The time CBF was below ischemic thresholds, in the present study, was not long enough to cause immediate infarction. However, it did not recover completely, and a relevant hypoperfusion persisted even hours after CPP had returned to near normal values. This observation most likely resembles the phenomenon of acute vasoconstriction after SAH (Bederson et al, 1998). Its persistence may constitute a reduced basis of CBF, which might be amplified by other factors. Occlusive hydrocephalus or hypoventilation can further elevate ICP, hypotension during anesthesia or temporary vessel occlusion during aneurysm surgery may further endanger appropriate perfusion. The sum of these factors may turn into hazardous focal or global perfusion deficits.
Brain Tissue Oxygenation
Changes of brain tissue oxygenation have been shown to correlate closely to changes of CBF and to changes of blood oxygenation (Menzel et al, 1999). As storage capacity for oxygen in brain tissue is low and the metabolic demand is particularly high after critical events like ischemia or SAH, ptiO2 must be expected to react quickly on variations of CBF. When LCBF decreased and recovered in the first hour after SAH, ptiO2 behaved alike in the present experiments. After 2 h, ptiO2 had fully recovered to baseline levels and continued to rise above baseline levels despite persisting hypoperfusion.
As tissue oxygenation is a resultant of supply and consumption of oxygen, our results indicate a reduction of oxygen utilization when blood supply has recovered enough for sufficient oxygen delivery. This finding supports the hypothesis of a metabolic depression and an uncoupling of CBF and oxidative metabolism and is in accordance with data about cerebral perfusion and metabolism gathered previously by patient studies (Carpenter et al, 1991; Martin et al, 1984; Jakobsen et al, 1990; Voldby et al, 1985). Carpenter et al performed early positron emission tomography studies in SAH patients before aneurysm surgery and observed a significant 25% decrease of CMRO2 even if patients had no signs of vasospasm in angiograms, suggesting a primary metabolic alteration and uncoupling of CBF and metabolism after SAH (Carpenter et al, 1991). However, in these studies, patients were not examined in the first hours after SAH.
Concerning the first hours after SAH, data are scarce and not unequivocal. If only the first 60 to 90 mins after SAH were considered, our findings are in accordance with the results of several previous studies. Critchley et al found an almost parallel fall and recovery of both CBF and ptiO2 during the first 60 mins after experimental SAH and concluded that changes of interstitial tissue oxygenation mirror changes of CBF after experimental SAH (Critchley and Bell, 2003). Prunell et al measured tissue oxygen tension (ptiO2) and LDF in rat brains after SAH. They found a rapid decrease of both parameters immediately after SAH and a concomitant, almost complete recovery in the following 90 mins. Simultaneously, CBF and CMRO2 were measured by autoradiography and determination of the arteriovenous difference of oxygen content. Both CBF and CMRO2 were markedly reduced 15 mins after SAH while there was no difference compared with sham-operated rats after 90 mins. The reduction of CMRO2 was not accompanied by an increase of oxygen extraction as should be expected if a depression of CMRO2 was secondary to a primary decrease of CBF. The authors, thus, concluded that the coupling of CBF and cerebral metabolism was maintained and that the metabolic depression was the primary event followed by a decrease of CBF (Prunell et al, 2004).
Our experiments did confirm neither a near-complete recovery of CBF nor a parallel course of CBF and tissue oxygen tension. CBF did not recover to baseline, and the decrease of ptiO2 was less pronounced than the decrease of CBF in the first 120 mins and even exceeded baseline values thereafter. Negative correlations between the final ptiO2 value and CPP and LCBF in the first minutes after SAH and a positive correlation between the final ptiO2 and LCBF values 3 to 6 h after SAH support the idea that the primary impact of hemorrhage is the trigger for a depression of CBF and oxygen utilization and an uncoupling of both. Even if oxygen supply is well restored after several hours, it might not be useful as a substrate for tissue metabolism. This is in accordance with patient studies mentioned previously (Carpenter et al, 1991; Martin et al, 1984; Voldby et al, 1985; Hayashi et al, 2008). Furthermore, the present experiments suggest that depression of oxygen utilization starts very early after SAH and persists for the following hours.
The pathophysiological cascade behind this phenomenon is not clear. Changes of oxygen metabolism seem to be more distinct, with more severe neurologic deficits and with larger amounts of subarachnoid blood (Grubb et al, 1977; Jakobsen et al, 1991). If normal circulation is timely restored after cerebral ischemia or hypoperfusion, the failure of vital cell functions may be avoided (Heiss, 1992).
In contrast to ischemia, the sudden and distinct reduction of CBF in SAH is not followed by an appropriate reperfusion but rather by a persistent perfusion deficit, which recovers gradually and only incompletely. After the onset of a perfusion deficit, the brain will first attempt to extract more oxygen, but this capacity is limited. As excitatory transmitters are released and cells are depolarized, the metabolic demand increases while, at the same time, the supply of oxygen is still limited. Lack of oxygen, lactacidosis, and increased energy demand may ultimately result in cellular and mitochondrial calcium overload and mitochondrial failure (Fiskum, 2000; Zauner et al, 2002). Failure of mitochondrial function has been observed in experimental SAH (Marzatico et al, 1988) and might play a crucial role for the development of secondary brain injury (Sehba and Bederson, 2006; Siesjo et al, 1999).
A deficient oxygen utilization as indicated by the present results may be due to a persistent mitochondrial dysfunction and results in an accumulation of oxygen in the brain. Although this was not studied in these experiments, the particular configuration of surplus tissue oxygen combined with mitochondrial dysfunction theoretically could be the basis for an increased generation of reactive oxygen species as mitochondria—due to their oxidative capacity—are a major source of reactive oxygen species and this pathway of reactive oxygen species formation can be enhanced in cases of mitochondrial dysfunction (Lewen et al, 2000; Fiskum, 2000).
The present data gives information about the dynamics of pathophysiological disturbances in the first 6 h after experimental SAH. At the end of the observation time, CBF and ptiO2 reached a plateau. The ultimate duration and possible resolvement of perfusion deficits and excess tissue oxygenation are not exactly known. Previous clinical studies, however, suggest that deficit perfusion and reduction of CMRO2 may persist for days after SAH (Carpenter et al, 1991).
It has to be kept in mind that the exact time-course of pathophysiological changes in animals may be different from humans, as well as there are large interindividual differences among the collective of SAH patients. However, a decrease of CPP has been observed in humans and rats as well as acute vasoconstriction, metabolic depression, and delayed vasospasm. It, therefore, is likely that the fundamental patterns of hemodynamic and metabolic changes found in animals reflect those in humans.
In summary, we observe a form of incomplete global ischemia in SAH that is not followed by appropriate reperfusion but rather by a prolonged and only slowly recovering hypoperfusion. In part, the course and intensity of the perfusion deficit are determined by acute vasoconstriction. After an initial phase of parallel decrease of CBF and tissue oxygenation, both parameters were uncoupled later, suggesting that once oxygen supply is restored, it cannot be metabolized properly and accumulates in the brain. A prolonged perfusion deficit by both acute vasoconstriction and deficient oxygen metabolization are likely to be relevant factors of secondary brain damage after SAH. Further studies are required to investigate the reasons for acute vasoconstriction, metabolic depression, and whether mitochondrial dysfunction in fact causes an enhanced production of reactive oxygen species. These pathologic features may all bear therapeutic potential in the acute phase after SAH.
Footnotes
None of the authors has a financial or other conflict of interest to declare. The study was not supported by public or private funding.