
Abstract
Hypertensive encephalopathy occurs when acute changes in blood pressure cause breakdown of the blood—brain barrier (BBB). Angiotensin II (Ang II) plays a role in this pathophysiology. We determined whether Ang II directly regulates endothelial cell function at the BBB. In BBB microvessel endothelial cells (MECs), the Ang II (100 nmol/L; 0 to 6 h) effects on permeability to 125I-albumin and transendothelial electrical resistance (TEER) were assessed. Angiotensin II (100 nmol/L) caused significant time-dependent changes in both 125I-albumin permeability (25%) at 2 h and TEER (−8.87 Ω·cm2) at 6 h. Next, MECs were pretreated with the Ang II type 1 (AT1) receptor blocker telmisartan (1 μmol/L) or the Ang II type 2 (AT2) receptor blocker PD123,319 (1 μmol/L) followed by treatment with Ang II (100 nm). Telmisartan completely inhibited the Ang II-induced increase in 125I-albumin permeability in MECs whereas PD123,319 had no effect. Using western blot analysis, we showed that MECs express AT1 receptors but not AT2 receptors. Treatment with Ang II (100 nmol/L; 0 to 6 h) also increased total protein kinase C activity. In contrast, Ang II had no effect on the expression of occludin, claudin 5, or actin. These results show that Ang II directly modulates transcytotic and paracellular permeability in BBB endothelial cells and could contribute to the pathophysiology of hypertensive encephalopathy.
Introduction
In the United States, it is estimated that one in three persons have hypertension, making it one of the most common extensive medical conditions in the United States (AHA, 2008). Prolonged hypertension is a risk factor for cerebrovascular, cardiovascular, retinal, and end-stage renal diseases, whereas acute increases in blood pressure can be life threatening, leading immediately to organ dysfunction and permanent damage (AHA, 2008; Poulet et al, 2006). Severe acute changes in blood pressure are categorized as hypertensive emergencies when they are associated with end organ damage (cardiovascular, cerebrovascular, retinal, or renal). The most life-threatening event of a hypertensive emergency is the development of hypertensive encephalopathy, which results from an opening of the blood—brain barrier (BBB; Cherney and Straus, 2002; Gardner and Lee, 2007; Vaughan and Delanty, 2000).
The pathophysiology underlying hypertensive emergency is poorly understood. Because of its rapid onset, it is suggested that in most cases an initial trigger plus the higher blood pressure activates a positive feedback loop, which stimulates the release of vasoactive mediators (Haas and Marik, 2006; Kitiyakara and Guzman, 1998). Activation of this feedback loop begins a vicious cycle, which leads to continuous organ damage and continuous increases in blood pressure (Haas and Marik, 2006; Kitiyakara and Guzman, 1998). There are many factors, which contribute to the pathophysiology of hypertensive emergency. These include mechanical stress, endothelial dysfunction, oxidative stress and damage with potential release of cytokines, prostaglandins and nitric oxide. The renin—angiotensin system (RAS) in particular plays a critical role in the initiation and maintenance of hypertensive emergencies and in target organ damage (Haas and Marik, 2006; Kitiyakara and Guzman, 1998; Stefansson et al, 2000; Sunder-Plassmann et al, 2002).
Angiotensin II (Ang II), the major effector molecule of the RAS, plays an important role in maintaining blood pressure and fluid homeostasis by its actions in both peripheral tissues and the central nervous system (CNS; de Gasparo et al, 2000; Phillips and Sumners, 1998; Sumners et al, 2002). The physiologic effects of Ang II occur via the G-protein-coupled Ang II type 1 (AT1) and Ang II type 2 (AT2) receptors. The AT1 receptor-mediated actions are the best characterized. In both peripheral tissues and the CNS, it is evident that the AT1 receptor couples to a wide variety of signaling pathways (de Gasparo et al, 2000; Phillips and Sumners, 1998; Sumners et al, 2002). However, the classic AT1 receptor-mediated signaling pathway in both the periphery and the CNS is activation of phospholipase C with subsequent increases in diacylglycerol and intracellular Ca2+, leading to activation of protein kinase C (PKC) and Ca2+/calmodulin-dependent protein kinase II (de Gasparo et al, 2000; Phillips and Sumners, 1998; Sumners et al, 2002).
During a hypertensive emergency, there is an uncontrolled upregulation of the RAS. More Ang II is released, and Ang II leads to vasoconstriction, increased blood pressure, increased volume contraction, naturesis, and subsequent ischemia. These responses can result in hypoperfusion of the kidney, which in turn, will stimulate the release of renin to produce still more Ang II. Additionally, Ang II can stimulate the release of other vasoactive and hypertensive compounds such as vasopressin, aldosterone, and catecholamines as well as stimulate the production of proinflammatory cytokines. The latter can themselves mediate opening of the BBB (Haas and Marik, 2006; Kitiyakara and Guzman, 1998; Zhang et al, 2000).
The BBB forms a metabolic and physiologic regulatory interface between the peripheral circulation and the brain extracellular fluid. The BBB can be modulated by the crosstalk, which occurs between brain endothelial cells, astrocytes, pericytes, microglia, and neurons. This intercellular interaction has led to the concept of the neurovascular unit (NVU) where each cell type in the NVU contributes to BBB function (Hawkins and Davis, 2005). At the BBB, a lack of vesicular transport limits transcytosis (vesicular-mediated permeability across cells), whereas tight junctions (TJs) obliterate intercellular spaces and so eliminate paracellular permeability (passage between cells). The net result is a barrier with a high transendothelial electrical resistance (TEER; ∼2,000Ω·cm2), which primarily reflects paracellular permeability, and with little unrestricted leakage of proteins, which primarily reflects transcytosis (Hawkins and Davis, 2005).
Many studies have investigated the effect of hypertension on cerebral microvessels and have shown that hypertension causes changes in the cerebrovasculature, including alterations in BBB physiology. These effects of hypertension on the BBB have been attributed in part to the mechanical stresses that occur with increased pressure (Grubb and Raichle, 1981; Kaya et al, 2003; Kucuk et al, 2002; Oztas and Sandalci, 1984). However one study showed that inhibition of the AT1 receptor blocks the hypertension-related increases in cell permeability and cerebral edema even in the absence of lowered blood pressure (Ito et al, 2000). This suggests a role for the RAS and Ang II in modulating BBB function. Early studies using bovine brain microvessel endothelial cells (MECs) showed that treatment with Ang II or an Ang II agonist stimulated an increase in fluid phase endocytosis (Guillot and Audus, 1990, 1991a; Guillot et al, 1990). Finally, it has been shown that Ang II itself undergoes AT1 receptor-mediated transcytosis in BBB endothelial cells (Rose and Audus, 1998, 1999). This transcytosis is predominately from the apical (blood) to basolateral (brain) surface, thus allowing other cells of the NVU to be exposed to Ang II not only from the CNS but also the circulation.
However, the direct effects of the RAS, specifically Ang II, on BBB or brain MEC function have not been fully investigated. Such studies are vital if one is to differentiate the indirect effects of hypertension itself from the mediators of hypertension. As many of the actions of the humoral mediators likely evolved to be adaptive to events such as hypotensive crises and are pathologic only when activated inappropriately, it is a great advantage to be able to study their mechanisms of action in the absence of changes in blood pressure. In these studies, we investigate the direct effects of Ang II on cultured monolayers of BBB MECs and investigate possible cellular mechanisms underlying Ang II-mediated changes in BBB function.
Materials and methods
Preparation of Mouse Brain Microvessel Endothelial Cells
Brains were removed from male CD-1 mice (Charles River Laboratories, Wilmington, MA, USA) and cerebral MECs isolated using a modified method of Banks et al (2004). Briefly, microvessels were isolated from mouse cortical gray matter using a sequence of enzymatic digestions and differential centrifugations. Cells were suspended in culture medium (Dulbecco's Modified Eagle's media/F-12 nutrients, horse serum (20%), penicillin, streptomycin, bFGF (1 ng/mL), and puromycin (4 μg/mL)) and seeded on collagen/fibronectin-coated dishes. Three days after the initial plating, cells were then fed with culture medium without puromycin and fed every two days afterwards. Cells were passaged one time and seeded on either collagen/fibronectin-coated dishes or Transwell filters at 50,000 cells/cm2. The confluent cells were used between days 7 to 10 after passage.
Preparation and Application of Angiotensin II, Peptides, and Drugs
The Ang II, pharmacologic agents and peptides were dissolved in appropriate solvents and then serially diluted in phosphate-buffered saline (PBS) to the appropriate concentrations for treatment. All the drugs, peptides, and control solutions (PBS) were added directly to the dishes of cells and incubated for various times.
Iodination of 125I-Albumin
Bovine serum albumin (5 μg; Sigma, St Louis, MO, USA) was radioactively labeled with 125I (Perkin Elmer, Waltham, MA, USA) according to the chloramine T method. Labeled albumin was then purified on a column of G-10 Sephadex hydrated with 0.25 mol/L phosphate buffer containing 1% unlabeled bovine serum albumin. Phosphate buffer was used to elute the iodinated protein.
In Vitro Assay for 125I-Albumin
Flux of 125I-albumin across the MEC monolayer was performed to determine whether Ang II stimulated transcytosis. Briefly, MECs were seeded on 0.45 micron Transwells filters and allowed to grow for 7 days. After treatment, the growth media was replaced with assay buffer containing (in mmol/L) 141 NaCl, 4 KCl, 2.8 CaCl2, 1 MgSO4, 10 HEPES, 10 glucose, 1 K2HPO4, and 1% BSA for 30 mins at 37°C. 125I-albumin (5 × 106 c.p.m./mL) was added to the upper chamber (lumen). Immediately, 100 μL of solution was removed from the upper chamber to determine the concentration of 125I-albumin in the donor chamber. Samples (500 μL) were removed from the lower chamber (abluminal) at times 15, 30, 60, and 90 mins and replaced with fresh assay buffer. The amount of radioactivity in each sample was determined and flux was calculated using the following equation:
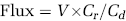
where V is the volume in the receiver chamber (0.6 mL), Cd is the concentration of 125I-albumin in the donor chamber at time 0, and Cr is the concentration of 125I-albumin in the receiving chamber at time T, with the Cr for times 30 to 120 being corrected for the amount of radioactivity remaining in the chamber from the previous time point. Flux was then plotted versus time and the apparent permeability (PSapp) calculated using linear regression analysis. The permeability of the membrane (PSmembrane) without cells was also calculated. The permeability of the cells (PScells) was calculated using the following equation
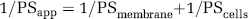
The permeability coefficient is calculated using the equation, PScells/SA, where SA is the surface area of the monolayer (0.33 cm2).
Transendothelial Electrical Resistance Measurements and Calculations
Transendothelial electrical resistance measurements were taken using an EVOM voltmeter and STX-2 electrode (World Precision Instruments, Sarasota, FL, USA). All TEERS are expressed as a change from pretreatment conditions.
Protein Kinase C Activity Assay
Total PKC activity in treated MECs was measured using the Non-radioactive Protein Kinase Assay kit from Calbiochem (San Diego, CA, USA). The protocol established by the company was used to measure PKC activity. Briefly, MECs were washed in ice-cold PBS and the cell extraction prepared using an extraction buffer containing (in mmol/L) 50 Tris-HCl, 5 EDTA, 10 EGTA, 50 β-mercaptoethanol, 10 benzamidine, and 1 PMSF, pH 7.5. Samples were sonicated and centrifuged according to the protocol. Protein concentration in the supernatant was determined using the Coomassie Plus—the better Bradford assay kit (Pierce, Rockford, IL, USA). Samples were then incubated with peptide pseudosubstrate followed by incubation with biotinylated antibody 2B9 and horseradish peroxidase-conjugated streptavidin. Aborbance was read at 492 nanometers. The absorbance is directly proportional to the amount of PKC activity and is corrected for the amount of protein in each sample.
Protein Analysis and Western Blot Analysis
Microvessel endothelial cells were washed in ice-cold PBS and protein was extracted using a RIPA buffer containing (in mmol/L) 150 NaCl, 50 Tris HCl, 1% NP-40, 0.5% Na deoxycholate, and 0.1% SDS. Protein concentrations were determined using the BCA Protein Assay kit (Pierce). SDS—polyacrylamide gel electrophoresis and western blot analysis were performed as described previously (Fleegal et al, 2005). Briefly, proteins were separated on 4% to 12% bis-tris gels (Invitrogen, Carlsbad, CA, USA) and transferred to nitrocellulose. The AT1 and AT2 receptors were detected by chemiluminescence (Pierce) using rabbit anti-AT1 (1:500), rabbit anti-AT2 (1:500), and HRP-conjugated anti-rabbit (1:10,000) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Occludin and claudin 5 were detected using mouse anti-occludin (1:2,000) and mouse anti-claudin 5 (1:2,000) antibodies (Invitrogen) and HRP-conjugated anti-mouse (1:10,000; Santa Cruz Biotechnology). Actin was run as a control to ensure that protein was loaded in equal amounts and that it was not degraded.
Lincoplex Analysis of Cytokine Release
Cytokine release into the media by MECs was assayed using the mouse cytokine/chemokine multiplex technology of the LINCOplex Biomarker Immunoassay kit (Millipore, St Charles, MO, USA). The protocol established by the company was used. Briefly, after treatment, media were collected from the cells and centrifuged to remove cell debris. Cytokine controls, standards, and samples were incubated with antibody-immobilized beads for the cytokines/chemokines overnight at 4°C followed by incubation with cytokine/chemokine detection antibodies at room temperature for 1 h. Cytokines were detected using streptavidin phycoerythrin and quantified using the Luminex200. Regression analysis was used to determine the concentration of cytokines in the media. This kit was used to assay for the following 22 cytokines: IL1α, IL1β, IL2, IL4, IL6, IL10, IL12, GM-CSF, IFNγ, TNFα, IL5, IL7, IL9, IL13, IL15, IL17, GCSF, IP-10, KC, MCP1, MIP-1α, and RANTES.
Results
Angiotensin II Alters Blood—Brain Barrier Endothelial Cell Permeability
In the first series of experiments, we determined the effects of Ang II on TEER, a measurement of paracellular permeability, and 125I-albumin transport, a measure of transcytosis, in isolated BBB endothelial cells. After treatment with Ang II (100 nmol/L; 0 to 6 h), there was a time dependent increase in 125I-albumin transport (25%) with the maximum increase seen at 2 h (Figure 1A). Additionally, there was a time dependent decrease in TEER with the maximum seen 6 h after Ang II treatment (Figure 1B). These results suggest that Ang II-mediated changes in 125I-albumin transport and TEER may involve different regulatory mechanisms. In the remaining experiments, we focused on investigating the mechanisms involved in the effects of Ang II on 125I-albumin transport at 2 h and TEER at 6 h. However it is important to note that when we looked at the 2 h time point only, Ang II also caused a significant decrease in TEER as well as the significant increase in 125I-albumin transport (Figure 2).
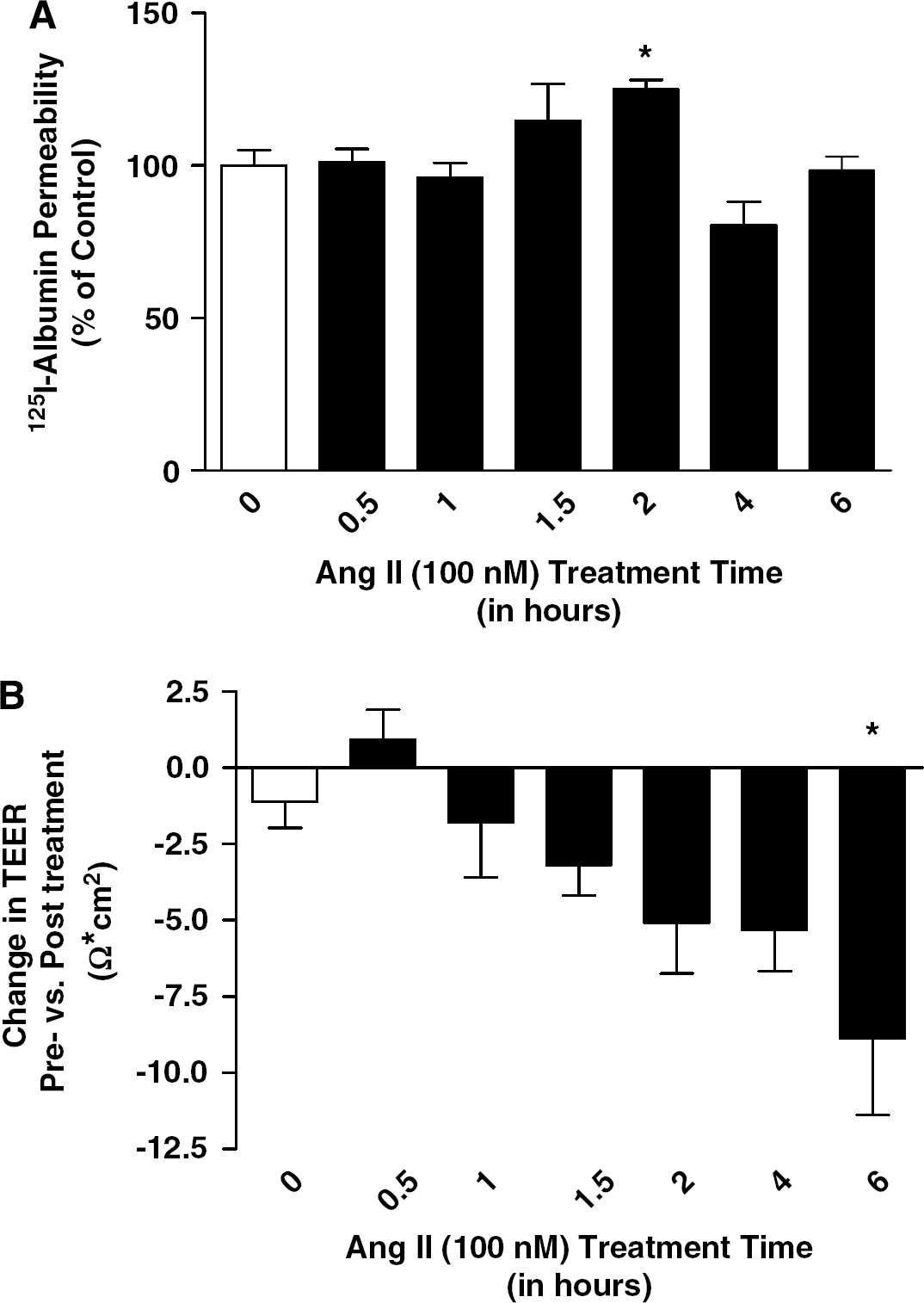
Ang II treatment increases 125I-albumin permeability and decreases TEER in a time-dependent manner in cultured BBB MECs. Confluent monolayers of cultured MECs were exposed to either PBS (0) or Ang II (100 nmol/L) for 0.5 to 6 h and permeability to 125I-albumin (
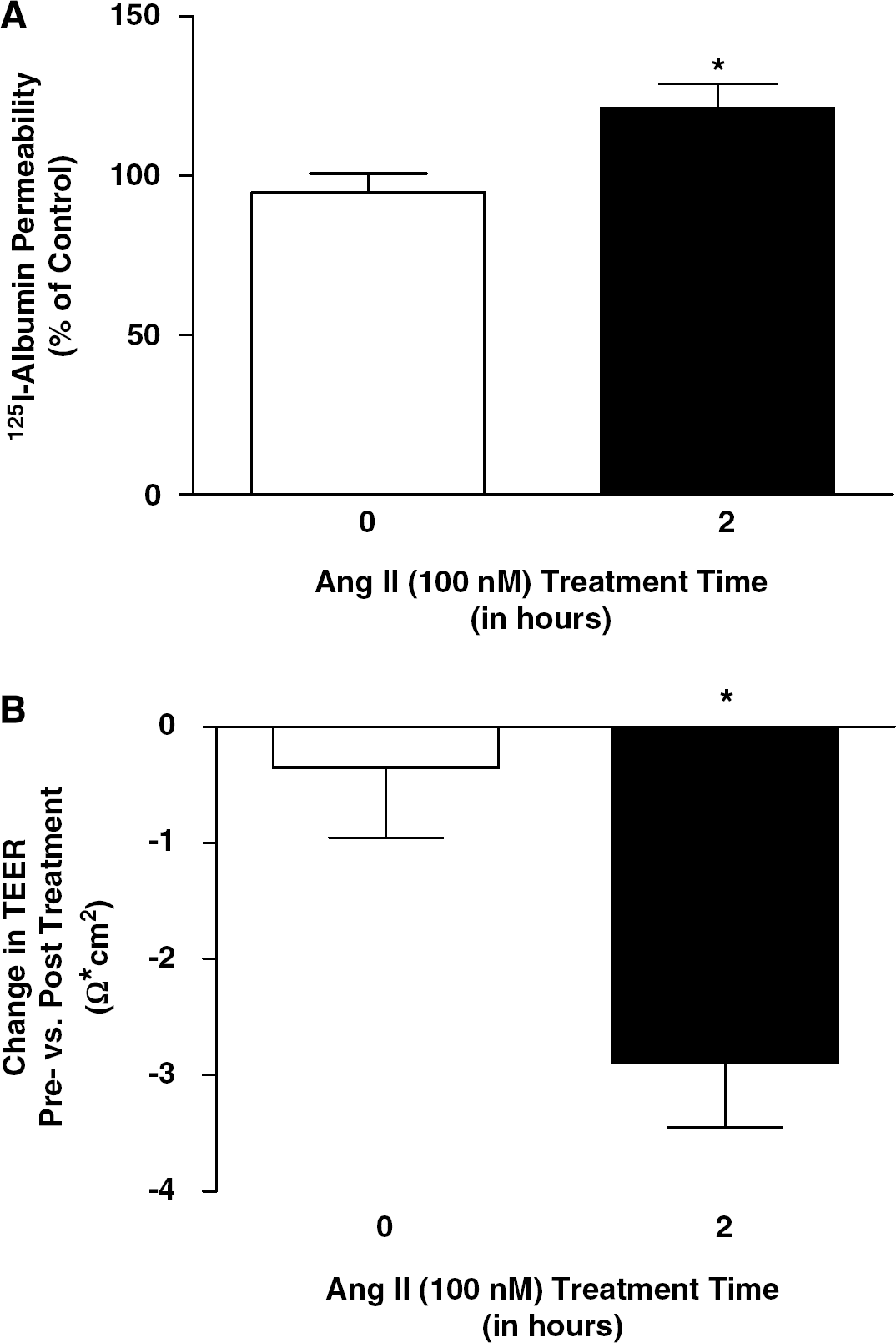
Ang II alters BBB permeability in cultured endothelial cells. Cultured brain MECs were exposed to either PBS (0) or Ang II (100 nmol/L) for 2 h and change in 125I-albumin permeability (
Angiotensin II-Mediated Changes in 125I-Albumin Transport Involve the Angiotensin II type 1 Receptor
We next determined whether the changes in 125I-albumin transport and TEER involved the AT1 or the AT2 receptor. For this set of experiments, cultured MECs were pretreated with either the AT1 receptor blocker, telmisartan (1 μmol/L; 15 mins) or the AT2 receptor blocker, PD123,319 (1 μmol/L; 15 mins) followed by Ang II (100 nmol/L) for 2 h. Treatment with telmisartan significantly attenuated the Ang II stimulated increase in 125I-albumin transport whereas the AT2 receptor blocker PD123,319 had no effect on 125I-albumin transport (Figure 3). Inhibition of the AT1 receptor reduced the change in TEER at 6 h but this effect was not statistically significant (data not shown).
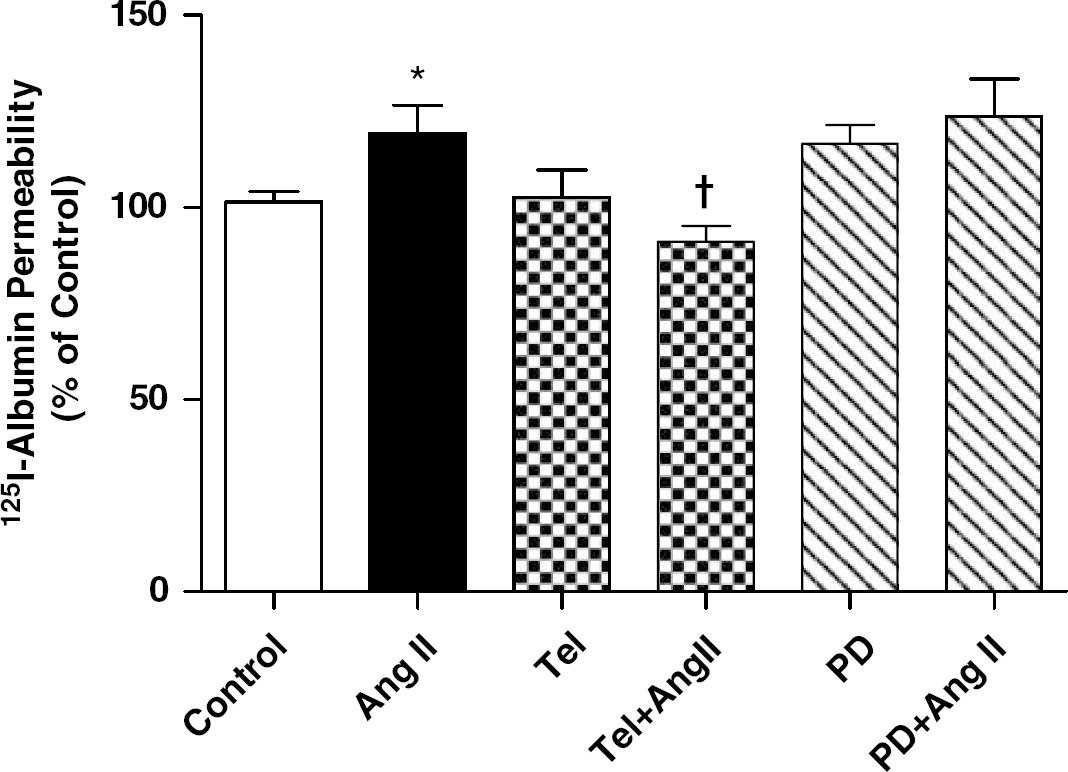
Inhibition of the AT1 receptor and not the AT2 receptor attenuates the Ang II-induced changes in 125I-albumin permeability. Confluent monolayers of cultured MECs were pretreated with either PBS, telmisartan (Tel; 1 μmol/L) or PD123,319 (PD; 1 μmol/L) for 15 mins and then exposed to Ang II (100 nmol/L) for 2 h. After the treatment paradigm, permeability to 125I-albumin was assessed. Bar graph shows the mean±s.e.m. 125I-albumin permeability (expressed as % of control). ∗P<0.05 compared with control; †P<.05 compared with Ang II (n=8 to 18).
As there is conflicting evidence on whether both the AT1 and AT2 receptors are expressed in BBB endothelial cells (Zhou et al, 2006), the next set of experiments was performed to determine which of these receptors are present on cultured BBB MECs. Protein was isolated from cultured brain MECs and from whole mouse brain, a control known to express both the AT1 and the AT2 receptors. Western blot analysis showed that whole mouse brain expressed both Ang II receptors (Figure 4). On the contrary, cultured MECs expressed only the AT1 receptor and not the AT2 receptor (Figure 4). This suggests that all actions of Ang II in BBB endothelial cells occur via the AT1 receptor.
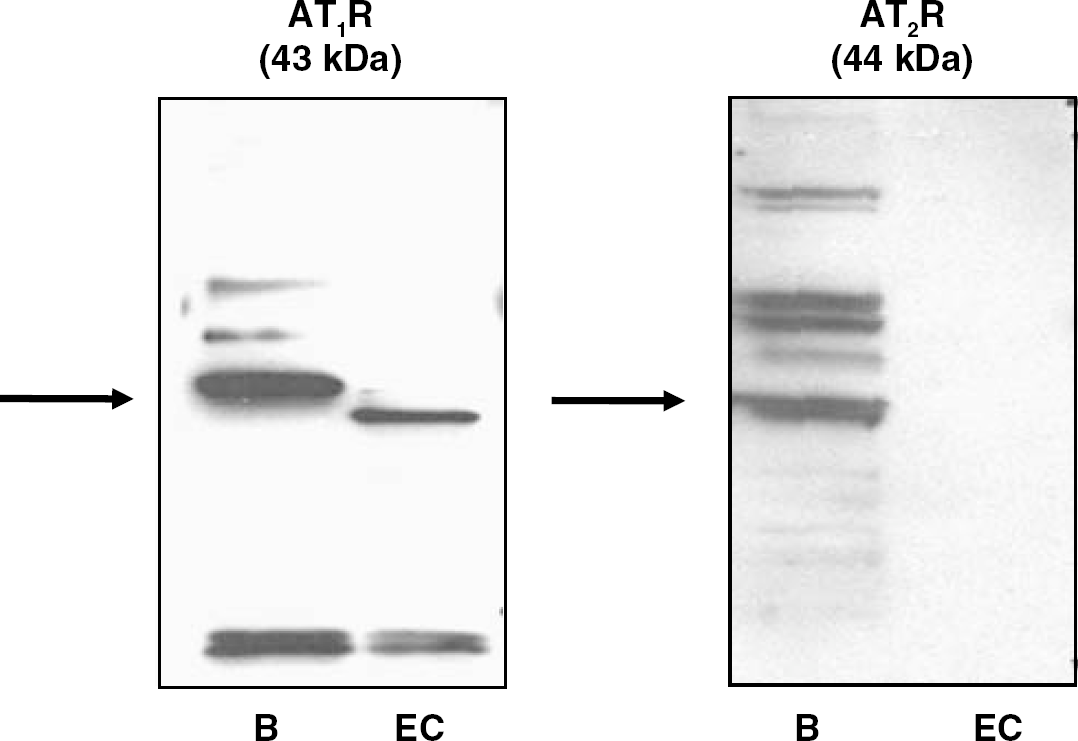
Cultured mouse brain microvessel endothelial cells express the AT1 receptor but not the AT2 receptor. Protein expression was evaluated using western blot analysis in whole brain (B) and mouse brain microvessel endothelial cells (EC).
Angiotensin II Stimulates Protein Kinase C Activity in Cultured Microvessel Endothelial Cells
In the next series of experiments, the intracellular mechanisms altered by Ang II in BBB endothelial cells were investigated. As discussed previously, many effects mediated by Ang II and the AT1 receptor occur via activation of the PKC pathway (Sumners et al, 2002). We next determined whether Ang II stimulates PKC activity in cultured brain MECs. In BBB endothelial cells treated with Ang II (100 nmol/L) for 0 to 6 h, there was a significant increase in PKC activity seen as early as 1 h, which remained elevated at both 2 and 6 h (Figure 5).
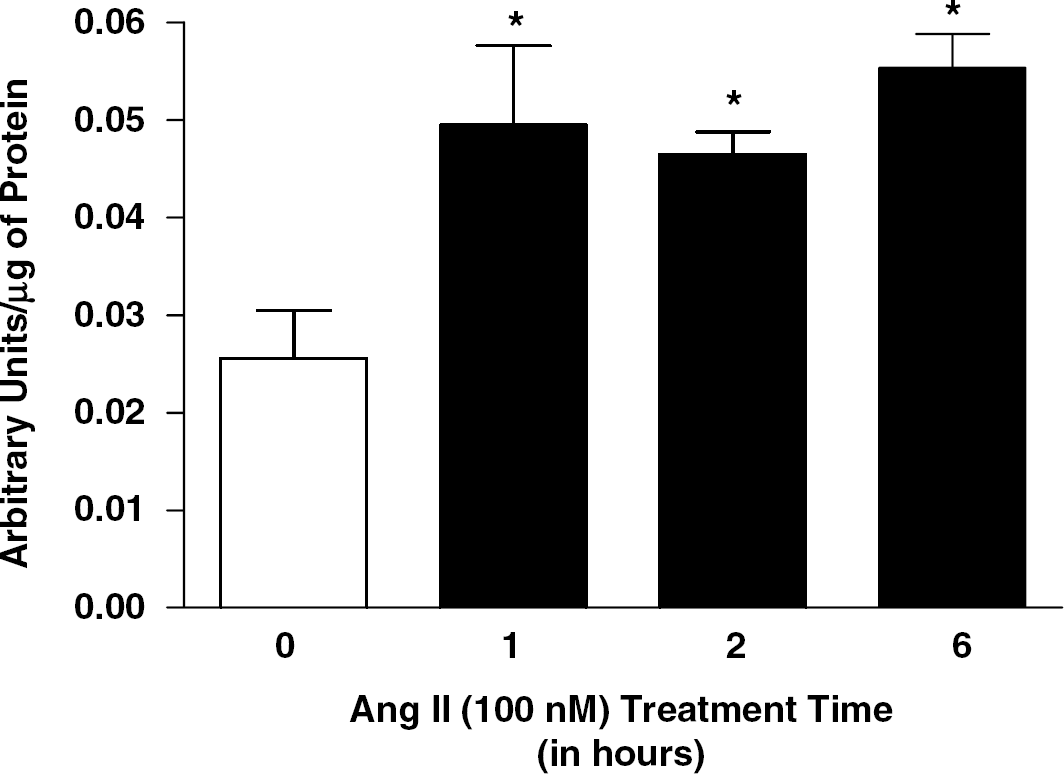
Ang II stimulates PKC activity in mouse BBB endothelial cells. Confluent monolayers of cultured MECs were exposed to either PBS (0) or Ang II (100 nmol/L) for 1 to 6 h and total PKC activity was measured as described in the methods. Bar graph shows the mean±s.e.m. PKC activity. ∗P<0.05 compared with control (0) (n=5 to 8).
As studies have shown that PKC can regulate paracellular permeability and several studies have suggested that PKC regulates the TJs (Clarke et al, 2000; Fleegal et al, 2005; Wosik et al, 2007), we also looked at whether Ang II altered the expression of the TJ proteins occludin and claudin 5. There was no change in the expression of these two proteins or the cytoskeletal protein, actin (Figure 6; Table 1). Finally, we looked at the effects of Ang II on the release of 22 different cytokines. There was no significant change in the release of these cytokines at either the 2 or 6 h time points (data not shown).
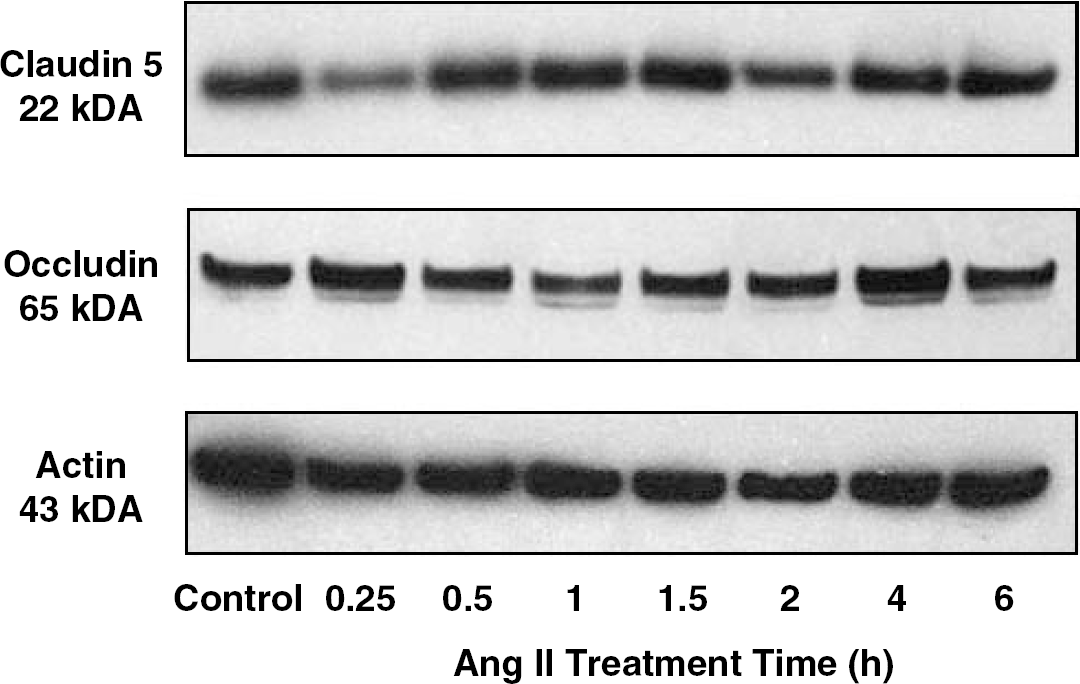
Ang II does not alter expression of claudin 5, occludin or actin. MECs were treated with PBS (0) or Ang II (100 nmol/L) for 0.25 to 6 h and protein expression was evaluated using western blot analysis. Representative western blots showing the expression of claudin 5, occludin, and actin.
Protein expression of claudin 5, occludin, and actin is not regulated by Ang II in vitro
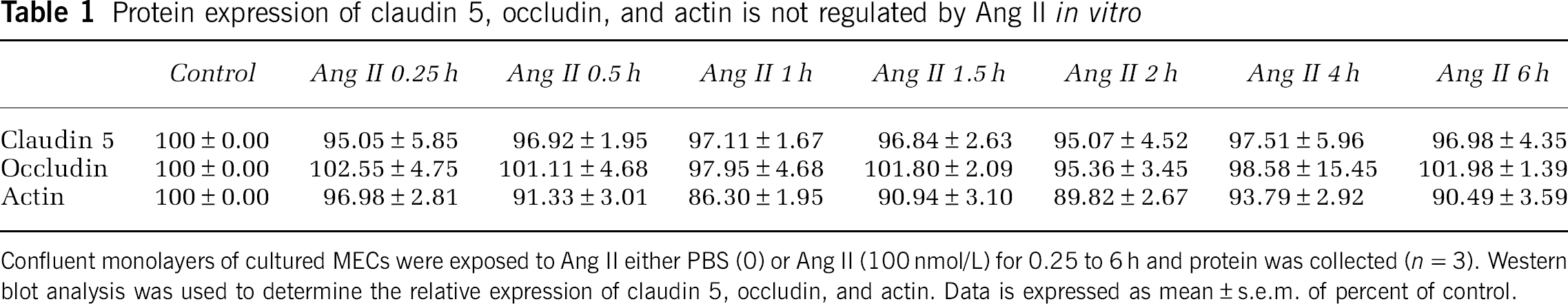
Confluent monolayers of cultured MECs were exposed to Ang II either PBS (0) or Ang II (100 nmol/L) for 0.25 to 6 h and protein was collected (n=3). Western blot analysis was used to determine the relative expression of claudin 5, occludin, and actin. Data is expressed as mean±s.e.m. of percent of control.
Discussion
The results presented here show that Ang II modulates permeability of BBB endothelial cells. These effects are presumed to involve the AT1 receptor but not the AT2 receptor for two reasons. (1) The AT1 receptor blocker telmisartan inhibited the Ang II stimulated increase in 125I-albumin transport whereas the AT2 receptor blocker had no effect on the Ang II-mediated increase in 125I-albumin transport. (2) Western blot analysis confirmed the presence of the AT1 receptor but not the AT2 receptor in cultured brain MECs. These studies have also begun to investigate the intracellular signaling mechanisms involved in the Ang II-modulated changes in both transcellular and paracellular permeability. These studies have shown the activation of PKC in cultured brain MECs before, and sustained throughout, the Ang II-mediated effects on cellular permeability.
Although these studies are not the first to suggest that Ang II plays a role in modulation of the BBB, they are the first to directly link Ang II to changes in BBB endothelial cell permeability. Early in vivo studies have suggested that Ang II does modulate changes in the cerebrovasculature. For example, monkeys showed an increase in 15O-water in the brain after intracerebroventricular injection of Ang II, indicating an increase in cerebrovasculature permeability (Grubb and Raichle, 1981). Additionally, studies in rat showed that intravenous injections of Ang II, which cause acute hypertension, caused a reversible increase in BBB permeability to Evans blue albumin (Oztas and Sandalci, 1984). However, it is difficult to determine if these were direct effects of Ang II or indirect effects because of mechanical stresses from increased blood pressure and flow. Therefore, it is a great advantage to be able to study the mechanisms of action of Ang II in the absence of changes in blood pressure. The studies presented here are in agreement with early studies that used bovine brain MECs to show that treatment with Ang II, an Ang II agonist or the PKC activator, phorbol myristate acetate stimulates an increase in fluid phase endocytosis (Guillot and Audus, 1990, 1991a; Guillot et al, 1990). However, it is important to note that a recent study has shown that Ang II released from astrocytes contributes to establishing and maintaining BBB properties in vitro (Wosik et al, 2007). The main reason there could be a difference in results between this recently published study and our current study is the treatment paradigm. We were focused on studying the effects of increased Ang II in an already established cultured monolayer whereas the paper by Wosik et al (2007) was looking at the effects of Ang II on a monolayer of endothelial cells as it was growing and developing its BBB properties.
In addition to showing a role for Ang II in modulating BBB function, these studies also suggest that in our model system, Ang II mediates its physiologic effects through the AT1 receptor. Other studies have shown the presence of Ang II binding sites and the presence of both AT1- and AT2-receptor mRNA and protein in brain MECs (Guillot and Audus, 1991b; Ibaragi and Niwa, 1989; Zhou et al, 2006). However, only the AT1 receptor has been shown to colocalize with the endothelial cell marker Glut1 (Zhou et al, 2006). Our results confirm the presence of the AT1 but not the AT2 receptor in cultured BBB endothelial cells. Our results along with the literature suggest that the AT2 receptor is not expressed by the BBB endothelial cell but is expressed by other cells of the NVU (Gallinat et al, 2000; Zhou et al, 2006). These results would then suggest that any effects of Ang II on cultured BBB endothelial cells involve the AT1 receptor.
Finally, our studies have begun to investigate the intracellular signaling mechanisms regulated by Ang II. As stated earlier, classically, Ang II-mediated physiologic responses involve activation of the AT1 receptor, which leads to activation of PKC (Sumners et al, 2002). The studies presented here show that Ang II also increases PKC activity in cultured brain MECs. Whether Ang II stimulates activity of similar isoforms of PKC in endothelial cells as compared with other cells of the CNS and periphery remains to be determined. However, a prime candidate to be investigated is the PKCδ isoform. Inhibition of the PKCδ isoform has been shown to prevent BBB breakdown and protect against hypertensive encephalopathy (Qi et al, 2008).
Protein kinase C is not the only intracellular signaling mechanism capable of modulating the physiologic effects of Ang II. A recent study showed that Ang II via the AT1 receptor stimulates adhesion of platelets and leukocytes in the cerebral microvasculature (Ishikawa et al, 2007). Studies have also shown that antagonism of the AT1 receptor will reverse macrophage infiltration of the microvessels and decrease expression of IL1β and TNFα in SHR (Takemori et al, 2000; Zhou et al, 2005, 2004). Other studies have shown alterations in the distribution of the TJs in microvessels of the SPSHR (Ito et al, 2000; Lippoldt et al, 2000). We also looked at the effects of Ang II on changes in both TJ protein expression and release of cytokines in cultured MEC monolayers. We showed no change in TJ protein expression and the release of 22 cytokines, which included but was not limited to IL1β and TNFα. Further studies need to investigate in greater detail the cellular mechanisms used by Ang II to alter both transcellular (125I-albumin transport) and paracellular (TEER) permeability in BBB endothelial cells.
In conclusion, these studies show that Ang II directly modulates BBB endothelial cell function by altering both transcellular and paracellular permeability. Furthermore, these studies confirm the presence of the AT1 receptor and not the AT2 receptor in cultured brain MECs. This leads us to suggest that the effects of Ang II in this model system occur via activation of the AT1 receptor. The cellular mechanisms involved in the Ang II-mediated changes in BBB endothelial cell function are not quite clear. However, they may involve PKC because Ang II stimulates PKC activity in these cells. On the contrary, Ang II does not appear to regulate the expression of the TJ proteins nor does it seem to regulate the release of cytokines. These studies indicate that Ang II may directly regulate BBB function by altering transcellular and paracellular permeability and by stimulating PKC activity.