
Abstract
Diabetic retinopathy (DR) is the leading cause of vision loss as a major complication of diabetes mellitus. The blood—retinal barrier (BRB) breakdown is a critical early event in the pathogenesis of DR. It has been known that the rennin-angiotensin system (RAS) is important in the progression of the DR via angiotensin II (Ang II), the effector of RAS. In this study, we showed that blockade of Ang II attenuates vascular endothelial growth factor (VEGF)-mediated BRB breakdown in DR. In streptozotocin-induced diabetes, retinal vascular permeability increased with upregulation of VEGF, where Ang II and its receptors were upregulated. Ang II induced VEGF expression in retinal endothelial cells accompanied by loss of tight junction proteins. However, the blockade of Ang II by perindopril, an angiotensin converting enzyme (ACE) inhibitor, inhibited upregulation of VEGF, and prevented the loss of tight junction proteins. Moreover, inhibition of Ang II by perindopril attenuated increased vascular permeability of diabetic retina accompanied by recovery of tight junction proteins in retinal vessels. Therefore, we suggest that the RAS involves in increased vascular permeability during early stage of DR, which is mediated by VEGF. Furthermore, the ACE inhibitor may have a therapeutic potential in the treatment of diabetic BRB breakdown.
Keywords
Introduction
Diabetic retinopathy (DR) is one of the major complications of diabetes mellitus and the leading cause of vision impairment and blindness in the Western world (Frank, 2004). The major risk factors for progression of DR are the duration of diabetes, poor glycemic control, high blood pressure, and elevated serum lipids (Davidson et al, 2007). Diabetes alters the structure and function of most cell types including both retinal vasculature and neural functions, which induces the blood—retinal barrier (BRB) breakdown in the early stage of DR (Antonetti et al, 1999; Lorenzi and Gerhardinger, 2001). The BRB breakdown is a key early event in the pathogenesis of DR, which results in vascular leakage and the development of macular edema, which is the leading cause of vision loss in diabetic patients (Frank, 2004; Davidson et al, 2007; Cunha-Vaz et al, 1979). The BRB breakdown is characterized by vascular leakage because of increased vascular permeability.
The BRB is essential for normal function of the retinal microenvironment and low permeability (Kim et al, 2006). Like our previous reports, the cellular interactions, regulating blood—neural barrier by modulating both brain angiogenesis and tight junction formation (Lee et al, 2003), also are critical in retinal barrier genesis (Choi et al, 2007). The specific junction molecules in retinal endothelial cells are requisite for the maintenance of barrier function. Recently, we have shown that zonula occludens-1 (ZO-1) and occludin are well-characterized components of the tight junction in retinal endothelial cells (Choi et al, 2007; Kim et al, 2007a, 2007b). Zonula occludens-1 is a cytoplasmic protein which links occludin to the other intracellular junction structures, and is inversely related to permeability in BRB as well as that of occludin (Choi et al, 2007; Kim et al, 2007a, 2007b, 2008 [14]). Vascular permeability is defined as the movement of fluids and molecules between blood vessels and the underlying tissues, which results from the disruption of intercellular endothelial junctions. Vascular endothelial growth factor (VEGF), originally isolated as a vascular permeability factor, is the best-known proangiogenic stimulus increasing the vascular permeability of microvessels to circulating macromolecules through uncoupling of endothelial cell to cell junctions (Weis and Cheresh, 2005).
The rennin-angiotensin system (RAS) is important in the progression of the DR via angiotensin II (Ang II), the effector molecule of the RAS (Danser et al, 1989; Van Dyk et al, 1994). There are known to be two types of Ang II receptors, type 1 and type 2 receptors (AT1R and AT2R; de Gasparo et al, 2002). In the retina, Ang II could induce pathologic changes in the retinal vascular system through AT1R, which primarily mediates the Ang II signaling (Nagai et al, 2005), whereas signaling through AT2R may be differentially regulated from that of AT1R and possibly acts in an opposite manner (Fujiyama et al, 2001). Although the differential roles of AT1R and AT2R in endothelial cells still remain to be elucidated, it is certain that Ang II increases vascular permeability and augments angiogenesis (Nagai et al, 2005; Fujiyama et al, 2001).
Considering that all components of the RAS are present in the retina (Sato et al, 1993; Wagner et al, 1996), and that VEGF and prorenin increase in the vitreous of patients with DR (Danser et al, 1989; Aiello et al, 1994), there would be a close association between VEGF, the RAS, and increased vascular permeability. Furthermore, the progression of DR was significantly suppressed by angiotensin converting enzyme (ACE) inhibition (Chaturvedi et al, 1998), which was supported by reports that Ang II increases VEGF-induced angiogenic activity in retinal endothelial cells (Otani et al, 1998), and retinal neovascularization is prevented by blockade of the RAS (Moravski et al, 2000). However, the role of the RAS in retinal permeability has not been elucidated yet.
In this study, we showed that blockade of Ang II attenuates VEGF-mediated BRB breakdown in DR. In diabetic retina, vascular permeability increased with upregulation of VEGF, where Ang II, AT1R, and AT2R were also overexpressed. Ang II, the effector molecule of the RAS system, induced VEGF expression in retinal endothelial cells, which was accompanied by loss of tight junction proteins. However, the blockade of Ang II by perindopril, an ACE inhibitor, inhibited upregulation of VEGF, and effectively prevented the loss of tight junction proteins. Interestingly, inhibition of Ang II by perindopril attenuated increased vascular permeability of diabetic retina, which was accompanied by recovery of tight junction proteins in retinal vessels of diabetic retina. Therefore, in addition to antiangiogenic activity of the ACE inhibitor in retinal endothelial cells, we herein suggest that the RAS involves in increased vascular permeability during the early stage of DR, which is mediated by VEGF. Moreover, the ACE inhibitor may have a therapeutic potential in the treatment of diabetic BRB breakdown.
Methods
Mice
C57BL/6 mice were purchased from Samtako (Seoul, Korea). Care, use, and treatment of all animals in this study were in strict agreement with the ARVO statement for the Use of Animals in Ophthalmic and Vision Research. C57BL/6 mice were kept in standard 12-h dark—light cycles and approximately 23°C room temperature.
Cell Culture
Human retina microvascular endothelial cells (HRMECs) were purchased from the Applied Cell Biology Research Institute (Kirkland, WA, USA) and grown on attachment factor-coated plates in complete medium (Cell Systems, Kirkland, WA, USA) or in M199 medium supplemented with 20% fetus bovine serum, 3 ng/ml basic fibroblast growth factor (Millipore, Bedford, MA, USA), and 10 U/ml heparin (Sigma, St Louis, MO, USA). Human retina microvascular endothelial cells used in this study were taken from passages 4 to 6. Angiotensin II (150 μmol/L; AnaSpec Inc., San Jose, CA, USA) or perindopril, an ACE inhibitor (10 μmol/L; Sigma) treatment was performed in cells cultured in serum-free M199 supplemented with 1% (vol/vol) penicillin-streptomycin.
Induction of Diabetes in Mice
To induce diabetes, 10-week male mice were intraperitoneally injected with 180 mg/kg streptozotocin (Sigma). If plasma glucose concentrations was >300 mg/100 ml at 24 h after streptozotocin injection, mice were considered to be diabetic. To assess the antipermeable activity of an ACE inhibitor, perindopril, 10 μmol/L perindopril in 1 μl phosphate-buffered saline (PBS) or PBS only was intravitreously injected to diabetic mice of 7 days after streptozotocin injection.
Mouse Retinal Tissue Preparation
On 8 days after streptozotocin injections with or without intravitreal injection of an ACE inhibitor, perindopril (10 μmol/L/1 μl), mice were carefully killed and the eyes were enucleated and hemisected at the ora serrata. The number of mice used in each experimental group was six. The retinas were gently teased off the sclera using a fine brush. Contamination by retinal pigment epithelial cells was reduced to a minimum. Whole retinal proteins were extracted with lysis buffer (50 mmol/L Tris (pH 7.6), 150 mmol/L NaCl, 1% Triton X-100, 0.1% SDS, protease inhibitor cocktail (Sigma), 1 mmol/L PMSF) on ice for 20 mins, centrifuged at 14,000g for 20 mins, and then supernatants were harvested and stored at −80°C.
Leakage Assessment by Perfusion of Retinal Vessels with Fluorescein-Conjugated Dextran
Eight days after streptozotocin injection with or without intravitreal injection of an ACE inhibitor, perindopril (10 μmol/L/1 μl), deeply anesthetized mice were perfused through the tail vein with high molecular weight (MW=500,000) fluorescein-conjugated dextran (Sigma) dissolved in PBS. The number of mice used in each experimental group was six. After 1 h perfusion, the eyes were enucleated and fixed in 4% paraformaldehyde for 2 h. The retinas were dissected, flat-mounted in Dako mounting medium (DakoCytomation, Glostrup, Denmark), and viewed by fluorescence microscopy (BX50; OLYMPUS, Tokyo, Japan) at a × 400 magnification.
Western Blotting
Western blotting was performed using standard western blotting methods. The protein concentration was measured using a BCA protein assay kit (Pierce, Rockford, IL, USA). Equal amounts of protein were separated by electrophoresis on 5% to 10% SDS—PAGE and transferred electrophoretically onto nitrocellulose membrane (Amersham, Little Chalfont, UK). The membranes were blocked for 30 mins in 5% non-fat milk. The membranes after blocking were incubated overnight with anti-VEGF (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-Ang II (1:1000; Abcam Inc., Cambridge, MA, USA), anti-AT1R (1:1000; Abcam Inc.), anti-AT2R (1:1000; Abcam Inc.), anti-ZO-1 (1:1000; Zymed, San Francisco, CA, USA), or anti-occludin (1:1000; Zymed) at 4°C. After they were washed with PBS-T, the membrane was incubated for 1 h at room temperature with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) or anti-mouse IgG (1:10,000; Pierce) in PBS-T and 1% non-fat milk. To ensure the equal loading of protein in each lane, the blots were stripped and reprobed with an antibody against β-actin. Intensity values were normalized relative to control values. The blots were scanned using a flatbed scanner and the band intensity analyzed using the TINA software program (Raytest, Staubenhardt, Germany).
Immunocytochemistry
Zonula occludens-1 expression in intercellular junction was examined by an immunocytochemical method as our previous description (Min et al, 2005). Briefly, treated cells were fixed with 2% paraformaldehyde and permeabilized with 0.2% Triton X-100. After being washed in PBS, the slides were blocked with 3% bovine serum albumin for 1 h and the cells incubated with anti-ZO-1 (1:100; Zymed) at 4°C followed by incubation with anti-goat IgG-rhodamine (Santa Cruz Biotechnology). The slides were viewed by fluorescence microscopy (BX50; OLYMPUS).
Immunohistochemistry
The enucleated mice eyes used for immunohistochemistry were immersion fixed in 4% paraformaldehyde and subsequently embedded in paraffin. 4 μm-thick serial sections were prepared from paraffin blocks. Sections were deparaffinized and hydrated by sequential immersion in xylene and graded alcohol solutions, treated with proteinase K for 5 mins at 37°C and then treated with normal serum obtained from the same species in which the secondary antibody was developed for 10 mins to block nonspecific staining. Slides were incubated overnight at 4°C with anti-ZO-1 (1:100; Zymed) and anti-occludin (1:100; Zymed), followed by incubation with anti-goat IgG-rhodamine (Santa Cruz Biotechnology). The slides were viewed by fluorescence microscopy (BX50; OLYMPUS) and random images (five images per slide; five slides per eye; six different eyes) were digitized with a three charge-coupled device color video camera (IK-TU40A; Toshiba, Tokyo, Japan) and a frame grabber. Image-analysis software (Image-Pro Plus; Media Cybernetics, Silver Spring, MD, USA) was used to measure the area of positive immunofluorescence.
Statistical Analysis
Statistical differences between groups were evaluated using either a Mann—Whitney test or a one-way analysis of variance (ANOVA) with Tukey's post hoc test using SPSS for Windows, version 12. 0 (SPSS, Chicago, IL, USA). Mean±s.d. is shown. P0.05 was considered significant.
Results
Increased Vascular Permeability of Diabetic Retina is Accompanied by Increase of Ang II, AT1R, and AT2R as well as Vascular Endothelial Growth Factor
To investigate increased vascular permeability in the early stage of DR, fluorescein angiography using high molecular weight (MW=500,000) fluorescein-conjugated dextran was performed at 8 days after streptozotocin injection. In diabetic retina, fluorescein-conjugated dextran easily infiltrated through the vessel wall and diffused into the diabetic retina, which was recognized as diffuse fluorescence all around the retina. As shown in Figure 1A, the vascular permeability was upregulated in the diabetic retina compared with normal retina at 8 days after streptozotocin injection.
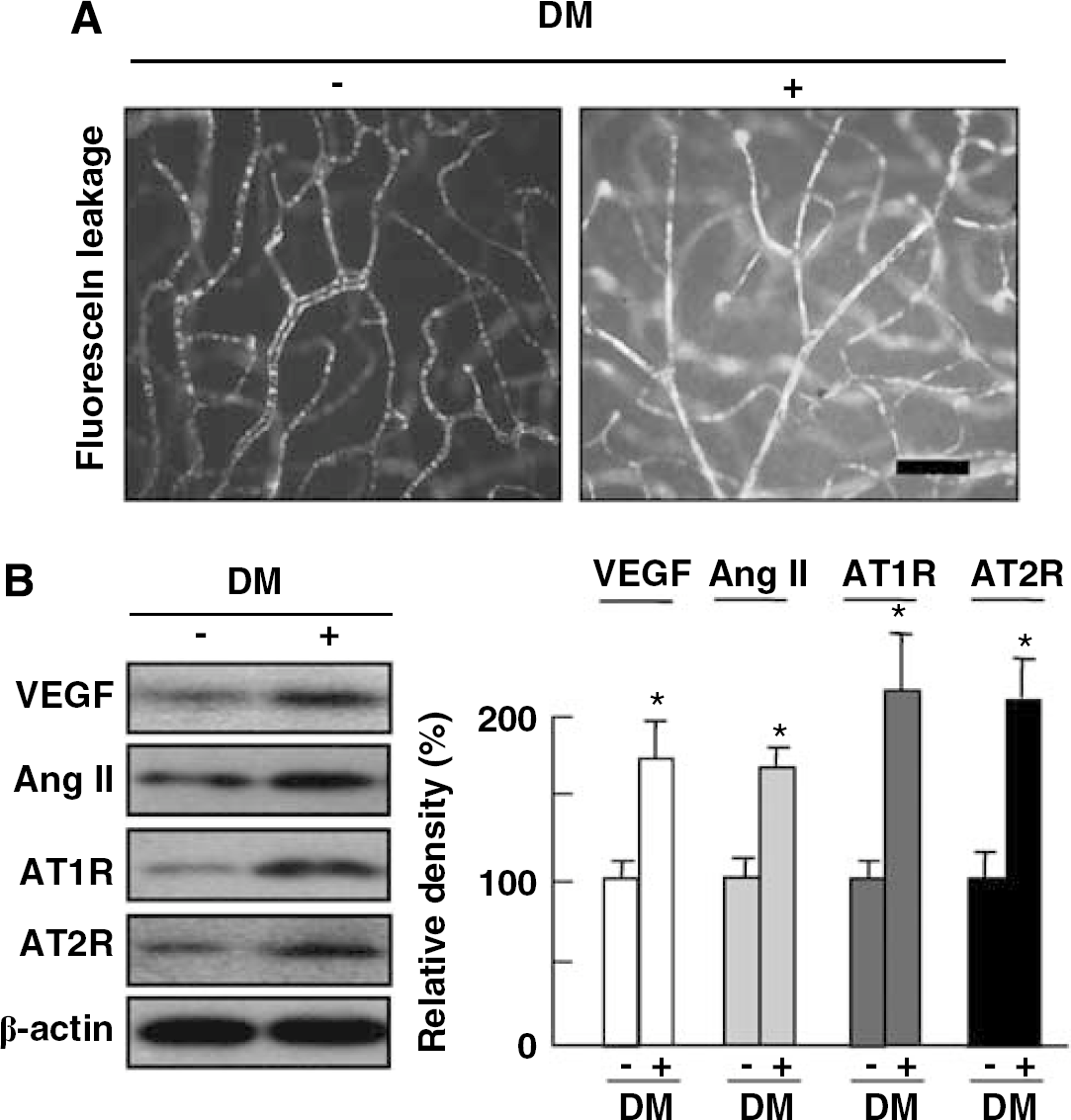
Increased vascular permeability of diabetic retina is accompanied by increase of Ang II, AT1R, and AT2R as well as VEGF. (
To evaluate the association between VEGF, the RAS, and increased vascular permeability in the early stage of DR, retinal proteins of diabetic mice were analyzed by western blot analysis using VEGF, Ang II, AT1R, and AT2R antibodies. As expected, VEGF expression was significantly increased by induced diabetes. With upregulation of VEGF, expression of Ang II, AT1R, and AT2R was all substantially increased in the diabetic retina than in control.
Angiotensin II Regulates Expression of Vascular Endothelial Growth factor and Tight Junction Proteins in Retinal Endothelial Cells
Based on the correlation between increased vascular permeability and loosening of the tight junctions in DR, we determined whether Ang II, the effector molecule of the RAS system, could regulate expression of VEGF and tight junction proteins in retinal endothelial cells. Human retina microvascular endothelial cells were incubated for 12 h with 150 μmol/L Ang II, or 10 μmol/L perindopril, an ACE inhibitor. It has been known that proangiogenic properties of Ang II might be because of the increase in VEGF and VEGF-receptor expression (Otani et al, 1998). Therefore, we checked VEGF expression after Ang II treatment in HRMECs. As shown in Figure 2A, Ang II induced VEGF expression in retinal endothelial cells, which were completely blocked by perindopril, an ACE inhibitor. Interestingly, with treatment of Ang II, ZO-1 was significantly reduced in HRMECs, which was prevented by perindopril treatment, whereas occludin did not show significant change compared with that in control or Ang II treatment (Figure 2A).
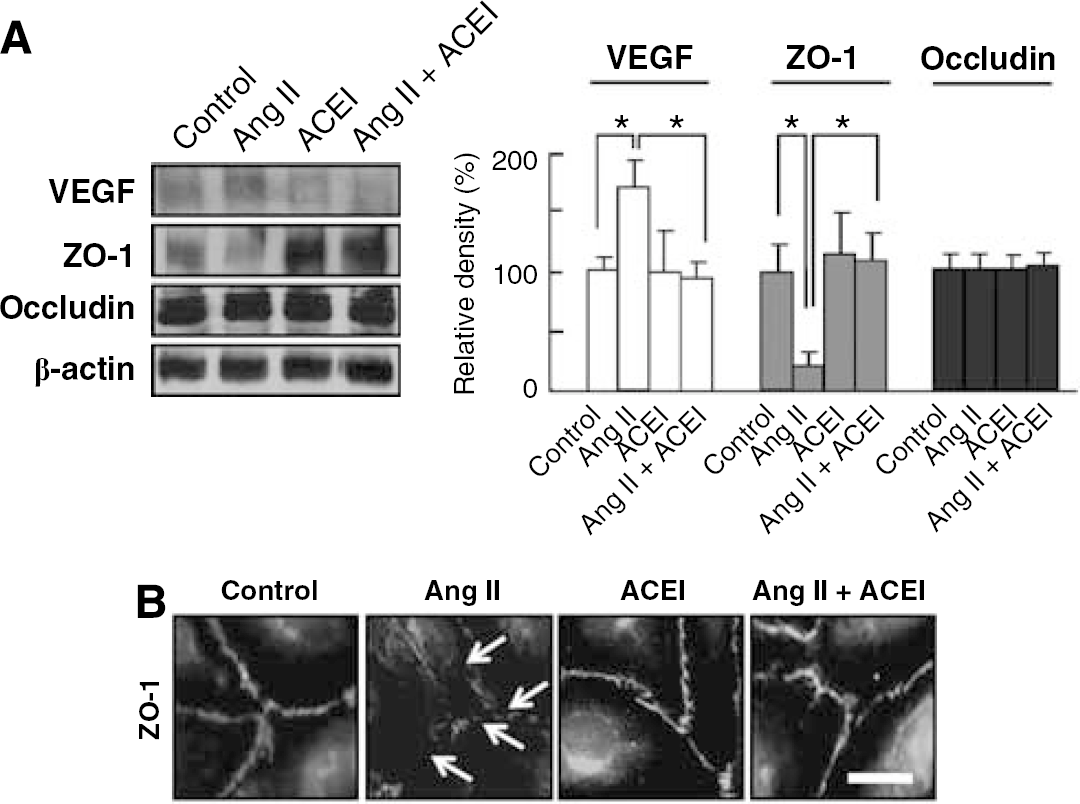
Ang II regulates expressions of VEGF and tight junction proteins in retinal endothelial cells. HRMECs were incubated for 12 h with 150 μmol/L angiotensin II, or 10 μmol/L perindopril, an ACE inhibitor. (
To confirm the effect of Ang II on expression of tight junction proteins in retinal endothelial cells, according to our previous reports (Choi et al, 2007; Kim et al, 2007a, 2007b, 2008), ZO-1 expression in intercellular junctions between cells was assessed with treatment of Ang II or perindopril. In confluent HRMECs, ZO-1 is a cytoplasmic protein well arranged along intercellular junctions. In Ang II-treated HRMECs, ZO-1 expression along intercellular junctions was markedly decreased, but cotreatment with perindopril completely blocked this effect (Figure 2B).
Blockade of Ang II Attenuates Increased Vascular Permeability of Diabetic Retina, which is Accompanied by Blockade of Loss of Tight Junction Proteins
To investigate the effect of blockade of Ang II on vascular permeability in diabetic retina, wholemount retinal preparation from 8 days after streptozotocin injection with intravitreal injection of perindopril, an ACE inhibitor (10 μmol/L/1 μl), was performed after 1 h perfusion of fluorescein-conjugated dextran. As shown in Figure 3A, perindopril markedly inhibited diffuse leakage from vessels in diabetic retina. To examine the effect of blockade of Ang II on restoration of tight junction proteins accompanying decrease of vascular leakage in diabetic retina, retinal proteins of 8 day after diabetes induction were analyzed for VEGF, ZO-1, and occludin. Upregulation of VEGF in diabetic retina was significantly blocked by perindopril. Although change of ZO-1 expression was more extensive than that of occludin, both ZO-1 and occludin significantly decreased in diabetic retina compared with control, which was recovered with intravitreal injection of perindopril (Figure 3B).
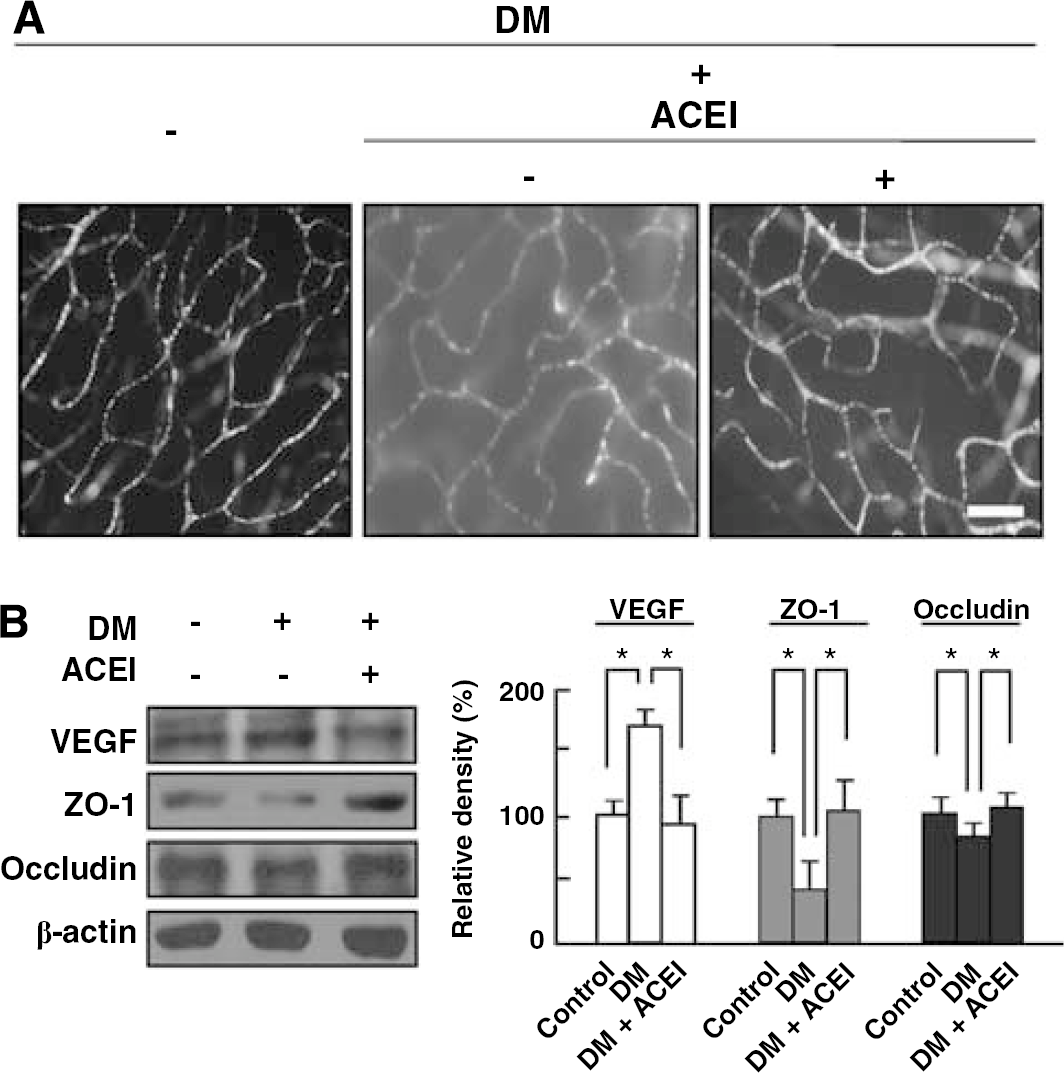
Blockade of Ang II attenuates increased vascular permeability of diabetic retina, which is accompanied by blockade of loss of tight junction proteins. (
Blockade of Angiotensin II Restores Loss of Tight Junction Proteins in Retinal Vessels of Diabetic Retina
To address whether blockade of Ang II could restore expression of tight junction proteins in diabetic retina, immunohistochemistry for ZO-1 and occludin was performed in diabetic retina with or without perindopril, an ACE inhibitor (10 μmol/L/1 μl). As shown in Figure 4, ZO-1 and occludin were strongly detected at the retinal endothelial cells of control mice, but these two important tight junction proteins were reduced by induction of diabetes (ZO-1 and occludin, 31±9% and 83±9%, respectively). However, with intravitreous injection of perindopril, diabetes-induced decrease in ZO-1 and occludin was restored (ZO-1 and occludin, 66±9% and 87±3%, respectively).
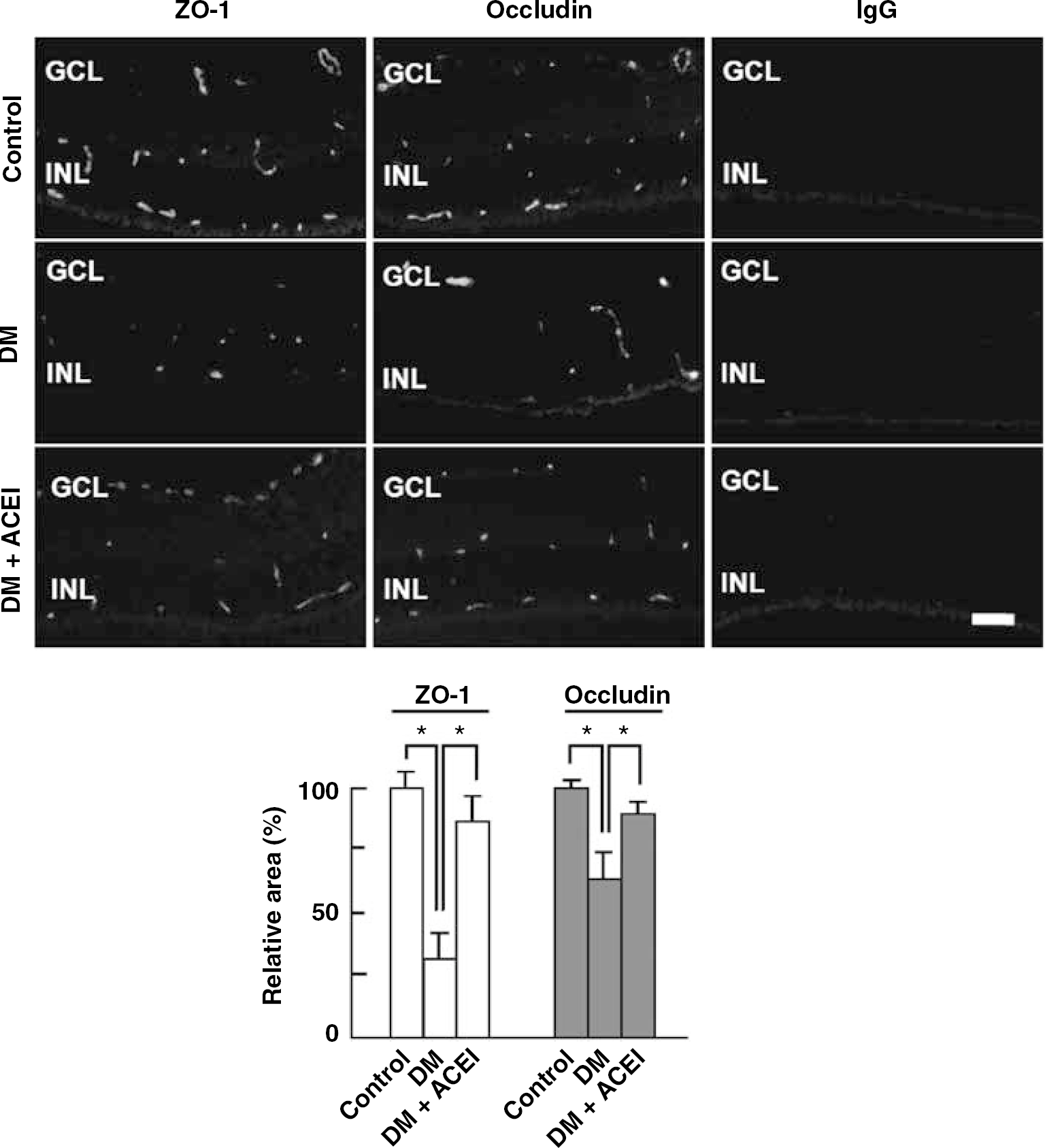
Blockade of Ang II restores loss of tight junction proteins in retinal vessels of diabetic retina. Immunohistochemistry for ZO-1, occludin, and IgG as a negative control was performed in diabetic retina with or without intravitreal injection of an ACE inhibitor, perindopril (10 μmol/L/1 μl). Figures were selected as representative data from six independent experiments. Scale bars: 100 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer.
Discussion
In this study, we showed that the RAS is involved in BRB breakdown in DR. In addition to previous reports for implication of Ang II in retinal angiogenesis and pathologic vascular growth (Otani et al, 1998; Moravski et al, 2000), we provided, for the first time to our knowledge, that blockade of Ang II attenuates VEGF-mediated BRB breakdown in DR.
Although differential roles should be further investigated, AT1R and AT2R have been found on retinal vasculature (Sarlos et al, 2003). In keeping with previous observations (Aiello et al, 1994; Chaturvedi et al, 1998), we showed that Ang II, AT1R, and AT2R as well as VEGF are upregulated in the condition of increased vascular permeability of diabetic retina. Vascular endothelial growth factor is crucial in the initiation and development of variable retinopathies (Aiello et al, 1994). In DR, VEGF per se is sufficient to induce vascular abnormalities including vascular leakage, microangiopathy, and neovascularization (Tolentino et al, 1996). Considering that Ang II increases VEGF and VEGF-receptor expression in retinal endothelial cells, and RAS blockade attenuates upregulation of VEGF in retinal neovascularization (Otani et al, 1998; Moravski et al, 2000), the RAS is also thought to be important in DR. Ang II acts through two types of receptors, AT1R and AT2R. The proangiogenic activity of Ang II is mediated via AT1R, in part, through activation of VEGF-related pathway (Tamarat et al, 2002). Besides proangiogenic activity, VEGF increases vascular permeability of microvessels to circulating macromolecules through uncoupling of endothelial cell to cell junctions (Weis and Cheresh, 2005). The BRB breakdown is a key early event in the pathogenesis of DR, which is characterized by vascular leakage because of increased vascular permeability. Taken together, vascular permeability in diabetic retina could be regulated by Ang II-induced VEGF expression.
We showed Ang II-induced VEGF enhancement lead to loss of tight junction proteins in retinal endothelial cells, which was inhibited by the blockade of Ang II by perindopril, an ACE inhibitor. In addition to experimental data, the therapeutic potential for RAS blockade has been suggested in the EUCLID study, where ACE inhibitor retarded the progression of proliferative DR (Chaturvedi et al, 1998), whereas ACE inhibition enhances ischemia-induced neovascularization in ischemic hind limbs (Fabre et al, 1999; Silvestre et al, 2001). Considering that ACE catalyzes the conversion to Ang II and the breakdown of bradykinin (de Gasparo et al, 2002), ACE inhibition could be mediated via inhibition of conversion to Ang II and, in part, via bradykinin accumulation. However, it has been known that bradykinin-related pathway is not involved in the ACE inhibition-induced prevention of microvascular disease in diabetic retina (Ebrahimian et al, 2005). Therefore, ACE inhibition could abrogate VEGF-mediated microangiopathy by blockade of Ang II. Taken together, it is reasonable to use agents interrupting RAS in the treatment of BRB breakdown of diabetic retina. In this study, we used perindopril as an ACE inhibitor. Originally, ACE inhibitor has been widely used as an antihypertensive agent. Recently, it has been also suggested that ACE inhibitors could inhibit angiogenesis and the growth of induced tumor (Volpert et al, 1996). In particular, perindopril, an ACE inhibitor, effectively retards tumor growth as well as angiogenesis at a clinically comparable dose without cytotoxicity (Yoshiji et al, 2002). This inhibitory effect is accompanied by the suppression of Ang II-mediated VEGF.
In summary, we showed that blockade of Ang II inhibits VEGF-mediated BRB breakdown in DR. Ang II, the effector molecule of the RAS, induced VEGF expression in retinal endothelial cells, which was accompanied by loss of tight junction proteins. Inhibition of Ang II by perindopril, an ACE inhibitor, attenuated increased vascular permeability of diabetic retina, which was accompanied by recovery of tight junction proteins in retinal vessels of diabetic retina. Antipermeable activity of ACE inhibition is therefore likely mediated by suppression of Ang II-mediated VEGF signaling. Therefore, our results suggest that Blockade of Ang II by ACE inhibitor could be an important therapeutic strategy for prevention of BRB breakdown in DR.