
Abstract
Matrix metalloproteinases (MMPs) have been implicated in the pathophysiology of ischemic stroke. In this study, we investigated the time course of gelatinolytic activation in a rat model of permanent ischemia. We observed an activation of MMPs as early as 30 mins after the ischemic insult, mainly in the nuclei of brain cells. Besides, we explored MMP-13 expression in brain samples of the animal model and stroke deceased patients. We observed an upregulation of active MMP-13 in rat brains (P< 0.05) after 90 mins of cerebral ischemia. Human infarct/periinfarct samples also showed higher levels of active MMP-13 (P< 0.05) compared with contralateral ones. Interestingly, we found that MMP-13 colocalized with 46-diamidino-2-phenyl indole signal by immunohistochemistry in both humans and rats, suggesting an intranuclear localization for MMP-13. Immunohistochemistry also revealed that MMP-13 was mainly produced by neurons, in both species, but also by oligodendrocytes in rats, and by astrocytes in humans. Finally we subjected a rat primary neuronal culture to oxygen and glucose deprivation (OGD) and we reproduced the nuclear translocation of MMP-13 in vitro. Nuclear extracts from cells confirmed upregulation of active MMP-13 after OGD (P< 0.05). These results suggest that MMP-13 activation and its nuclear translocation is an early consequence of an ischemic stimulus.
Introduction
Experimental and clinical studies have shown that ischemic brain injury after stroke is a dynamic process that evolves a period of hours to several days (Dirnagl et al, 1999; Sharp et al, 2000). This process includes several events such as oxidative stress, cell death, inflammation, as well as activation of endogenous adaptive and regenerative mechanisms (Baranova et al, 2007).
Matrix metalloproteinases (MMPs) are a family of zinc-binding proteolytic enzymes that normally remodel the extracellular matrix and pathologically degrade substrates as part of the neuroinflammatory response (Matrisian, 1990). Collagen and gelatin, as well as other extracellular molecules such as fibronectin, laminin, and a variety of proteoglycans can be degraded by MMPs (Yong et al, 1998). MMPs are either transmembrane or secreted proteins essential in the breakdown of the extracellular matrix around cerebral blood vessels and neurons. Thus, their action might lead to opening of the blood—brain barrier, brain edema, hemorrhage, and cell death (Jian Liu and Rosenberg, 2005).
Animal models of cerebral ischemia have reported that MMP-9 and also MMP-2 are abnormally expressed after an ischemic insult (Planas et al, 2001; Rosenberg et al, 1996). Indeed an increased in situ gelatinase/collagenase activity in brain samples from mice (Lee et al, 2004), rats (Justicia et al, 2003; Loy et al, 2002; Rivera et al, 2002), and humans (Rosell et al, 2006) has been reported after cerebral ischemia. Interestingly, mice lacking MMP-9 gene or treated with broad-spectrum MMP inhibitors exhibit diminished cerebral damage after stroke (Asahi et al, 2000).
A recent study reported by Amantea et al (2008), described an increased nuclear MMP-derived gelatinolytic activity in brain cells after reperfusion injury in a transient model of cerebral ischemia in rat, although their nuclear function remains unexplained yet.
Matrix metalloproteinase-13 (or collagenase-3) is a member of the collagenase subfamily of MMPs and has a central position in the MMP activation cascade. Its activation can be autoproteolytic or catalyzed by several MMPs (Leeman et al, 2002) and MMP-13 can degrade collagens but it also shows the highest gelatinase activity among collagenases (Knauper et al, 1996). Although MMP-9 has been widely studied in many animal models of cerebral ischemia as well as in human stroke, only a few investigations have reported the role of MMP-13 in the pathogenesis of brain ischemia.
To our knowledge, only a previous study has investigated MMP-13 after focal ischemia in rats reporting an increase in neurons, peaking at day 7 after reperfusion (Nagel et al, 2005). Besides, previous results from our group showed that high levels of MMP-13 are involved in infarct growth suggesting an early role in brain injury in stroke patients (Rosell et al, 2005).
The aim of the present study was to examine the time course of MMP-proteolytic activation and to determine MMP-13 expression after stroke. We observe an activation of MMPs as early as 30 mins after the ischemic insult in a rat model of permanent middle cerebral artery occlusion (pMCAO). Furthermore, among all the proteases involved in the process, we report evidence that active MMP-13 is upregulated in rats as well as in human postmortem brain after cerebral ischemia, mainly in neurons and oligodendrocytes. Interestingly, we observe that MMP-13 appears within the cell nuclei and that this effect is reproduced in vitro in rat cortical neuron cultures exposed to oxygen and glucose deprivation (OGD). The data presented suggest that MMP-13 activation and its nuclear translocation is an early consequence of an ischemic stimulus.
Materials and methods
Brain Tissue Samples
A total of 10 deceased patients (7 women and 3 men) who had an ischemic stroke within the previous 4 days (range, 12 to 96 h) were included in the study, demographic data are listed in Supplementary Table 1 online. On autopsy and during macroscopic exam, infarcted area was delimitated by an experienced neuropathologist (mainly through consistence and color of the parenchyma) and neuroradiology images were used to obtain brain tissue from ipsilateral, infarction (n = 10) or periinfarction (n = 7), and contralateral hemisphere (n = 10). All samples were obtained within the first 6 h after death and snap frozen in liquid nitrogen and stored at −80°C for Western blot or fixed with formalin for immunohistochemistry techniques. This study was approved by the Ethics Committee of the hospital and informed consent was acquired from relatives before the autopsy.
Experimental Stroke Model
All animal experiments were approved by the Ethics Committee of the Research Institute of the Vall d'Hebron Hospital and were conducted in compliance with the Spanish legislation and in accordance with the Directives of the European Union.
Infarction in the territory of the MCA was induced by vascular occlusion in Sprague—Dawley rats (Charles River Laboratories, Wilmington, MA, USA) weighing 280 to 310 g as described previously (Longa et al, 1989). Briefly, animals were anesthetized with isoflurane under spontaneous respiration. Body temperature was maintained at 37°C using a temperature controlled heating pad. The right common carotid artery was exposed through a midline incision in the neck and it was carefully dissected from the tissue using microsurgical technique. The occipital and superior thyroid branches of the external carotid artery were cauterized and the pterygo-palatine branch of the internal carotid artery was irreversibly occluded. The external carotid artery was tied and cut and then, a 4/0 nylon monofilament was passed via the external carotid artery and up the internal carotid artery 18 to 20 mm, blocking the MCA. To determine that the MCA was successful occluded, regional cerebral blood flow was measured in the cortex supplied by the MCA by continuous laser-Doppler flowmetry (Moor Instruments, Devon, England). At 1 day before stroke induction, a small burr hole in the right parietal bone (2 mm posterior and 3.5 mm lateral to bregma) was drilled leaving the dura intact, and a cannula was fixed to the skull with cyanacrylate and dental cement. Laser-Doppler flowmetry probe (0.5 mm diameter) was placed on the dura using the cannula and cortical regional cerebral blood flow was assessed during all surgery time to ensure MCA occlusion. Only animals with a regional cerebral blood flow reduction of more than 75% were included (n = 46). Sham-operated rats were subjected to the same surgical procedure without the occlusion of the MCA. Animals were killed at different times after artery occlusion (30, 60, 90, 120 mins or 48 h; n = 5 per group). One animal of the 48 h group died during the process. Finally, rats were deeply anesthetized and transcardially perfused with ice-cold saline. Afterward, both ipsilateral and contralateral brain hemispheres were rapidly dissected and kept frozen (–80°C) until use.
Analysis of Infarct Volume
To measure the infarct volume, animals were killed and brains were removed at 30, 60, 90, 120 mins or 48 h after MCAO and evaluated using 2,3,5-triphenyltetrazolium chloride staining (Bederson et al, 1986; n = 4 per group). The brain was sliced in 2-mm-thick sections and stained with 2% 2,3,5-triphenyltetrazolium chloride for 30 mins at room temperature followed by an overnight fixation with 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline. The image of the stained slices was captured using a CanoScan 4200F (Canon Inc., Lake Success, NY, USA) and the area of infarction and noninfarction tissue was outlined and quantified using an image analysis system (Scion Image version 4.02, Scion Corporation, Frederick, MD, USA). The infarct volume (corrected for edema) was calculated by integration of the lesion areas and expressed as a percentage of ipsilateral hemisphere.
Cell Culture
Cerebral cortical cells were obtained from fetal Sprague—Dawley rats at 17 days of gestation. After the meninges were carefully removed, cortices were enzymatically and mechanically dissociated as previously reported (Goldberg and Choi, 1993). Cells were resuspended in Basal Medium Eagle (Pan Biotech GmbH, Aidenbach, Germany) supplemented with 10% fetal bovine serum (Gibco BRL, Carlsbad, CA, USA), 10% FHS (Pan Biotech GmbH), 50U/mL penicillin, 50 µg/mL streptomycin (Gibco BRL), 2 mmol/L glutamine, and 30 mmol/L glucose (complete Basal Medium Eagle). Dissociated cells were seeded to 2.5×105 cells per milliliter and plated into poly-
Oxygen and Glucose Deprivation
Mature neuronal cultures (11 to 13 DIV, n = 10) were OGD by placing them in an anaerobic chamber (Invivo2, Ruskinn, Pencoed, UK), containing 5% CO2, 95% N2 at 37°C for 3 h with deoxygenated glucose-free Earle's balanced salt solution, containing 116 mmol/L NaCl, 5.4 mmol/L. KCl, 0.8 mmol/L MgSO4·7H2O, 1.0 mmol/L NaH2PO4·2H2O, 26.2 mmol/L NaHCO3, 1.8 mmol/L CaCl2·2H2O, 0.01 mmol/L glycine, pH 7.4. OGD was terminated by returning the cultures to the pre-OGD culture medium and normoxic conditions. Reoxygenation was performed for 24 h. Control neuronal cultures were exposed to balanced salt solution containing 5.5 mmol/L glucose, and kept in the standard incubator during the same time as OGD cultures. Cell viability was measured by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) reduction assay. After treatments, MTT (0.5 mg/mL) was added to cells and after 90 mins incubation at 37°C, the medium was replaced by dimethyl sulfoxide. The amount of formazan blue formed after MTT reduction was quantified spectrophotometrically at 560nm (Plumb et al, 1989).
Nuclear Extraction
Cell pellets were recovered with a rubber policeman in 0.5mL of cell lysis buffer 1 containing: 50 mmol/LTris HCl, pH 7.6, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% BRIJ-35, 0.02% NaN3, and 1% Triton X-100, 0.2 mmol/L phenylmethylsulfonyl fluoride, and 10 µg/mL aprotinin. Pellets were homogenized using a Dounce Homogenizer and centrifuged at 240g for 5 mins at 4°C. After 15 mins on ice, pellets were resuspended in 0.5mL 0.6% Nonidet P-40 lysis buffer. The homogenate was vortexed for 10 secs and centrifuged at 240g for 5 mins at 4°C. The supernatant was kept for analysis of the cytoplasmic fraction. Pellet was resuspended on 0.2mL of lysis buffer 2 containing: 20 mmol/L Tris HCl, pH 7.5, 125 mmol/L NaCl, 5 mmol/L MgCl2, 0.2 mmol/L EDTA, 12% glycerol, and 0.1% NP-40, sonicated and centrifuged at 20,000g for 15 mins at 4°C and the supernatant was kept for nuclear extract analysis.
To assess the purity of nuclear extracts, Western blots with anti-GAPDH as cytoplasm marker (1:1000, mouse, Ambion, Foster City, CA, USA) and anti-Histone H3 as nuclear marker (1:500, rabbit, Cell Signaling, Boston, MA, USA) were performed on 15% acrylamide gels.
Western Blot
Frozen samples were homogenized for Western blotting. Brain tissue (0.2 g) was mixed with 0.7mL of cold lysis buffer (50 mmol/L Tris-HCl, pH 7.6, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% Brij-35, 0.02% NaN3, and 1% Triton X-100), containing protease inhibitors (1 mmol/L phenylmethylsulfonyl fluoride and 7 µg/mL aprotinin) and centrifuged at 12,000g for 10 mins. Total protein content was determined by bicinchoninic acid assay (Pierce, Rockford, IL, USA). MMP-13 protein content was detected by Western blot in brain homogenates from rats (n = 25, 5 per group) and humans (n = 27, 7 to 10 per study area) as well as in the cell extracts (n = 5). Briefly, equal protein amount (30 µg in brain homogenates and 50 µg in cell extracts) was loaded in laemmli buffer with SDS-polyacrylamide gel electrophoresis (12%) at 100V. Separated proteins were transferred onto a PVDF membrane using a Transblot Cell (Bio-Rad, Hercules, CA, USA) during 1 h at 100V. Nonspecific bindings were blocked with 10% nonfat milk before membranes were incubated overnight at 4°C with mouse anti-MMP-13 antibody (Calbiochem, Gibbstown, NJ, USA) at 1:300 dilution. Secondary antibody goat anti-mouse-HRP (Chemicon, Billerica, MA, USA) was diluted 1:2000 and incubated at room temperature for 1 h. Before and after incubations, membranes were washed three times (10 mins each) with 0.05% Tween-phosphate buffer saline. The substrate reaction was developed with chemiluminescent reagent Immobilion (Millipore, Billerica, MA, USA) and visualized with a luminescent image analyzer (Las-3000, Fujifilm, USA).
Immunodetection of β-actin (Sigma-Aldrich, St Louis, MO, USA) was performed to verify that equal amounts of total protein were loaded for each brain sample.
In Situ Zymography
Brain frozen sections (12 µm) from rats were prepared using a cryostat (Leica CM3050S, Leica, Germany). Preparations were treated with the MMP fluorogenic substrate DQ-gelatin-FITC (Molecular Probes, Eugene, OR, USA) as described previously (Justicia et al, 2003). Some sections were treated with broad-spectrum MMP inhibitor 1,10-phenanthroline (10 mmol/L) to ensure specificity. Tissues were analyzed with laser confocal spectral microscope (Olympus FV-1000) and images were processed with Olympus Fluoview software (version 1.7a).
Immunohistochemistry/Immunocytochemistry
For immunohistochemistry assays, frozen sections (12 µm) were prepared using a cryostat. The following antibodies were used: rabbit anti-MMP-13 antibody (1:100; Sigma-Aldrich), mouse anti-NeuN antibody (1:100; Chemicon), mouse anti-glial fibrillary acid protein antibody (1:100; Sigma-Aldrich), mouse anti-Iba1 antibody (1:20; Abcam, Cambridge, UK), mouse anti-CNPase (1:200; AbD Serotec, Kidlington, UK). Briefly, sections were fixed in ice-cold acetone for 15 mins and then washed in Tween-phosphate buffer saline for 5 mins. The sections were then blocked with 10% bovine serum albumin (w/v)/0.3% Triton X-100 (v/v) in phosphate-buffered saline (blocking buffer) for 30 mins at room temperature, and then incubated overnight at 4°C with primary antibodies diluted in blocking buffer. After washing in Tween-phosphate buffer saline (3×5 mins), the sections were incubated with goat Alexa-Fluor 488 anti-mouse IgG or goat Alexa-Fluor 568 anti-rabbit IgG antibodies (Invitrogen, Carlsbad, CA, USA) diluted 1:500 in blocking buffer for 1 h at room temperature. Finally, the sections were thoroughly washed (3×5 mins) and then mounted on coverslips using Vectashield with 46-diamidino-2-phenyl indole (DAPI; Vector Laboratories, Burlingame, CA, USA).
For immunocytochemistry assay, cells were seeded and grown on glass coverslips. Fixation was performed with 4% paraformaldehyde for 20 mins and finally washed with Tween-phosphate buffer saline (3×5 mins). Blocking and antibody incubations were performed as described above.
Negative control sections received identical treatment except for the primary antibody. Tissues were analyzed with a fluorescence microscope (Olympus IX71) interfaced with an image analysis system. For colocalization studies, laser confocal spectral microscope (Olympus FV-1000) was used. Tissue images were processed with Olympus Fluoview version 1.7a software (Olympus, Japan). Three-dimensional reconstruction of tissue sections was performed using Imaris 4.5.2 (Bitplane AG, Switzerland).
Statistical Analysis
Data were analyzed using GraphPad Prism version 4.0 software. Kruskal—Wallis test was used followed by Dunn's Multiple Comparison test to assess intergroup differences. A P-value <0.05 was considered statistically significant at a 95% confidence level. Error bars in figures represent s.e.m.
Results
Gelatinase Activity Appears in Cell Nucleus as Early as 30 mins After Cerebral Ischemia
We assessed whether gelatinase proteolytic activity increased in brain parenchyma of rats that underwent a permanent focal cerebral ischemia by MCAO at short times (30, 60, 90 and 120 mins) and also 48 h after the ischemic insult.
Infarct size was measured to evaluate the intensity of ischemic injury of this model. 2,3,5-Triphenyltetrazolium chloride staining (Supplementary Figure 1 online) showed no lesion at 30 mins and a progressive increase of the infarcted tissue at longer times of ischemia yielding an extent infarction at 48 h after the occlusion (35.3 ± 15.6%, P < 0.01 versus 30 mins group; Supplementary Figure 1b online).
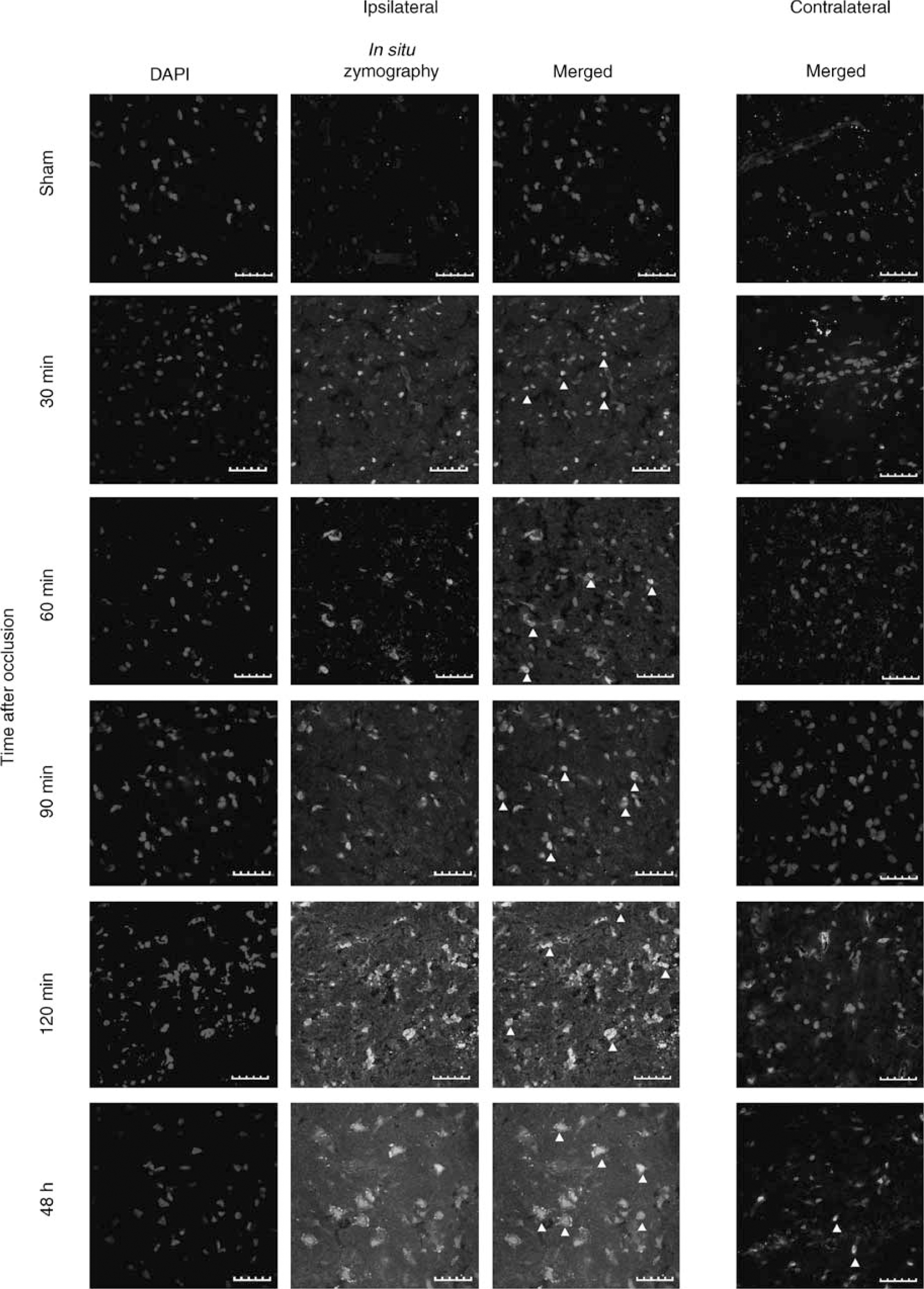
Images showing in situ zymography assay in striatum of rat brain preparations. Nuclei are stained with DAPI. In ipsilateral hemispheres proteolytic activity appears in nucleus (arrowheads) as early as 30 mins after MCA occlusion as seen in merged images. Contralateral hemisphere (merged images) only shows colocalization in few cells after 48 h of pMCAO. Scale bar = 40 µm.
During short times of ischemia (30 to 120 mins), gelatinase activation was detected mostly in the striatum (Figure 1) whereas at 48 h of brain ischemia gelatinolytic activity appeared also in the cortex.
With regard to the subcellular localization, gelatinase activity was already found in cell nucleus at 30 mins of brain ischemia (Figure 1) whereas at 90 and 120 mins after artery occlusion the gelatinolytic activity increased and appeared also in the cell cytoplasm. Gelatinase signal yielded the highest intensity at 48 h after MCA occlusion and could be found both in cell nucleus and cytosolic compartment (Figure 1). No proteolytic signal was found in sham-operated animals; however, a weak signal appeared in some nuclei in contralateral hemisphere at 48 h of cerebral ischemia. Sections treated with broad-spectrum MMP inhibitor 1,10-phenanthroline did not show any proteolytic activity (data not shown).
Active MMP-13 Protein Increases at Short Times of Cerebral Ischemia
Western blot experiments of rat brain homogenates showed an active MMP-13 form (MMP-13a) of 45 kDa (Figure 2A); however, no proMMP-13 form could be detected with this antibody. These experiments showed that MMP-13 protein level is elevated at all occlusion times when compared with sham animals. Furthermore, MMP-13a levels peaked in the 90 mins occlusion group (P < 0.05) and its presence remained overexpressed in the 48 h of permanent ischemia group (Figure 2B), although significant differences were not found.
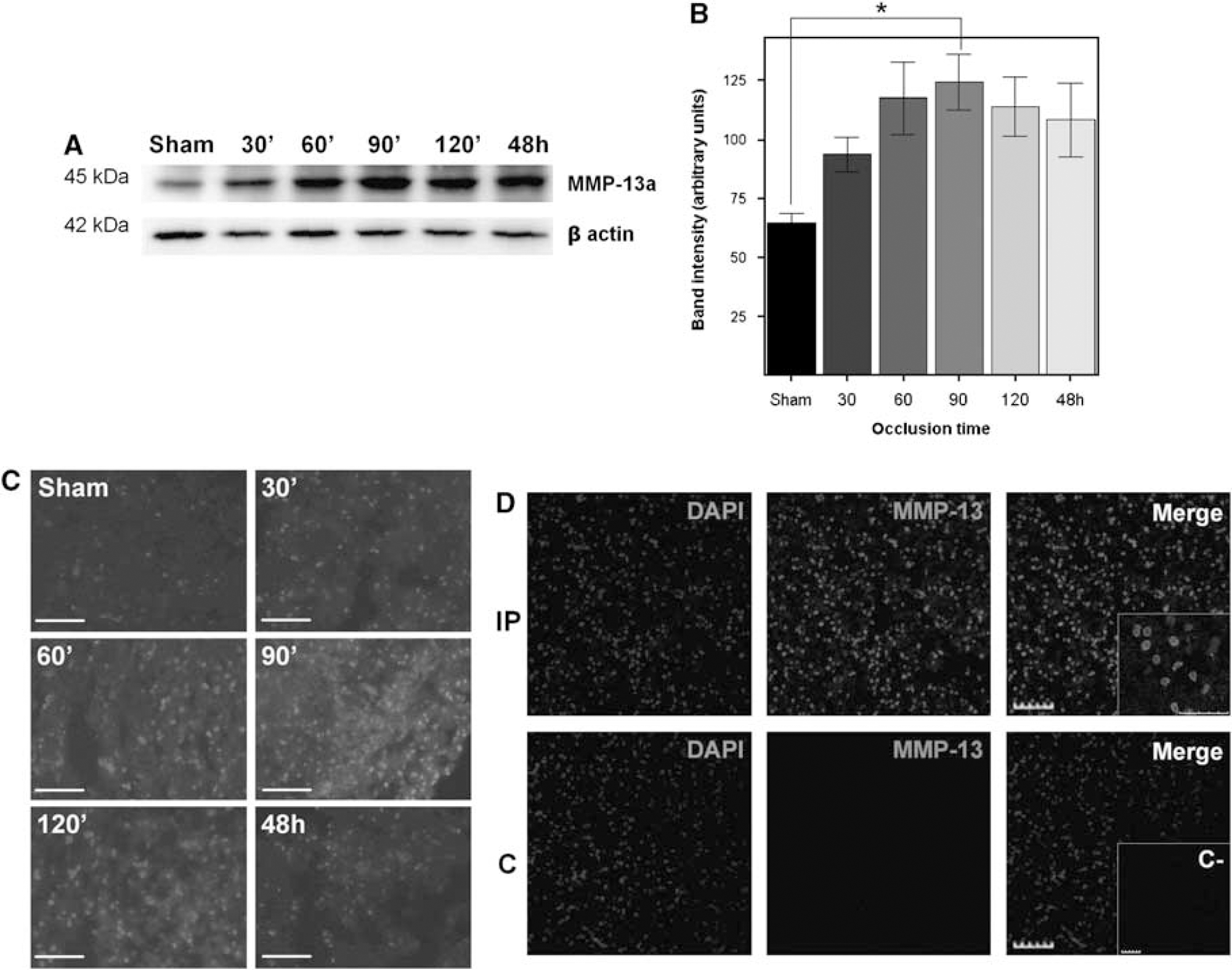
Changes in the expression of MMP-13 in rat brain after MCA occlusion. (
The immunohistochemical analysis of rat brain preparations showed consistent results because MMP-13 reactivity slightly increased in the ischemic tissue already after 30 mins of cerebral ischemia (Figure 2C) and peaked at 90 mins. Besides, the MMP-13 signal was mainly detected in the ipsilateral hemisphere whereas hardly any signal was discerned in the contralateral hemisphere (Figure 2D). It is worth noting that MMP-13 signal colocalized with DAPI signal within the infarcted hemisphere in many cells (Figure 2D insert).
Active MMP-13 Protein Level Increases in Human Brain After Stroke
We then examined MMP-13 protein expression in the brain tissue from deceased patients after an ischemic stroke. By Western blot, we were able to detect the MMP-13 proform (60 kDa, proMMP-13) as well as the cleaved active form of MMP-13 (48 kDa, MMP-13a; Figure 3A). Western blot analysis showed no differences for proMMP-13 among studied areas (Figure 3B). However, the infarct core, together with the periinfarct, showed the highest levels of MMP-13 active form (P < 0.05, Figure 3C) compared with contralateral areas. Immunohistochemistry confirmed that MMP-13 was upregulated within the infarction as well as in the periinfarct area (Figure 3D) and a low signal was displayed by the contralateral hemisphere preparations. Similarly to our observations in rats, human MMP-13 signal also colocalized with DAPI signal in both infarct and periinfarct areas in some cells (Figure 3D insert).
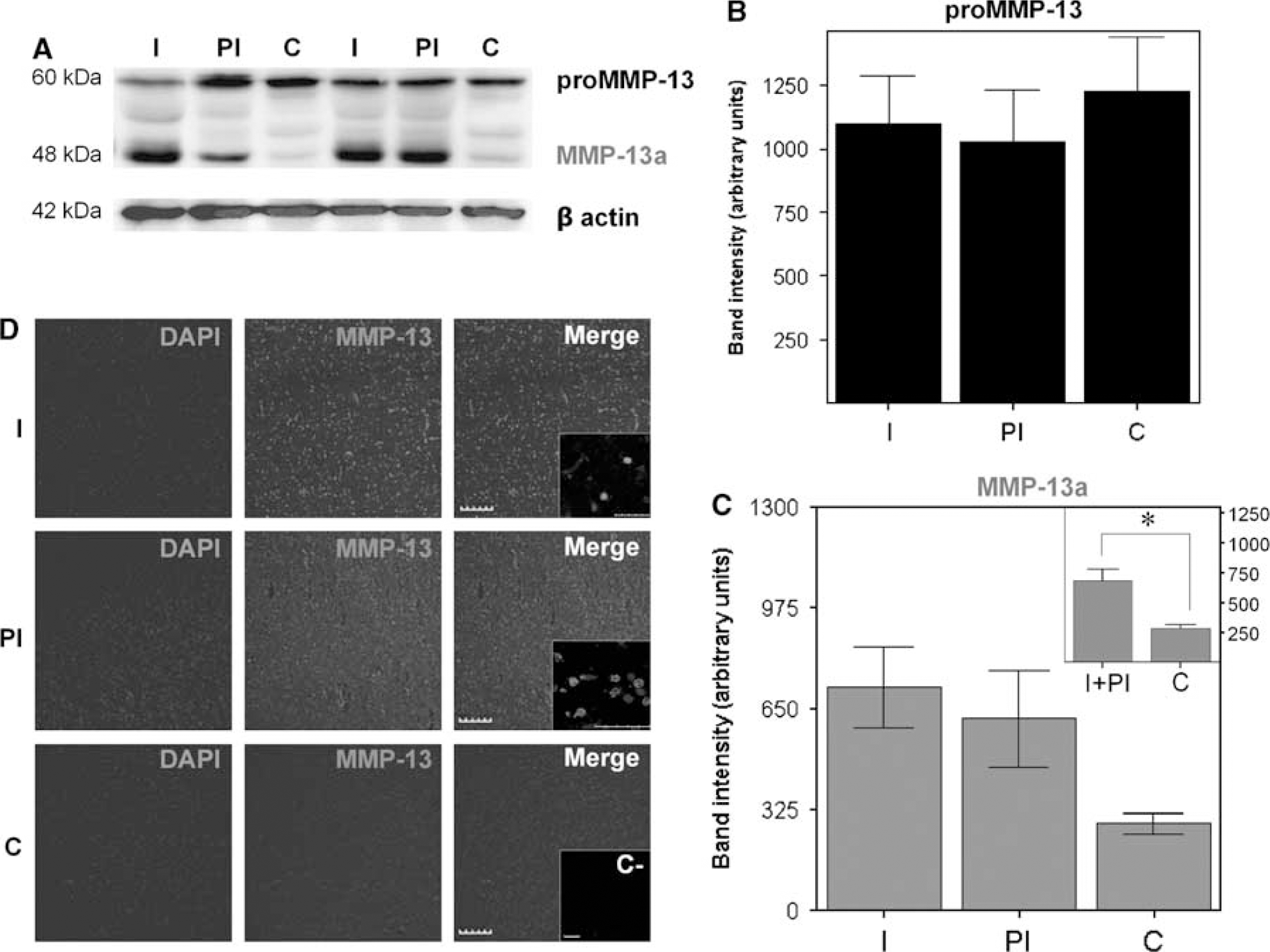
MMP-13 is upregulated in infarction/periinfarction of human stroke samples. (
MMP-13 is Found in Cell Nucleus After Stroke
To verify a possible nuclear localization of MMP-13, we performed a confocal analysis of both human and rat brain preparations. As in both set of samples, this colocalization could be neatly observed, we chose a rat brain preparation to illustrate this phenomenon because we were especially interested in early times of ischemia.
Confocal analysis showed colocalization of MMP-13 signal (in red, 568nm laser excitation) with DAPI nuclear staining (in blue, 405nm laser excitation) in rat preparations (Figure 4A). Further analysis from an amplified field in Figure 4A obtained with optical section separation of 1 µm (z interval) showed how MMP-13 immunoreactivity co-migrates with DAPI signal along the z interval (Figure 4B). Imaris software allowed us to create a three-dimensional reconstruction using the stack of 12 optical sections from the tissue. This rendition helped us to observe how the MMP-13 signal colocalize spatially with the DAPI signal in the infarcted brain tissue (Figures 4C and 4D).

Confocal analysis in a preparation of rat brain submitted to 90 mins of cerebral ischemia. (
MMP-13 is Found in Neurons and Oligodendrocytes in Rats
As MMP-13 appeared upregulated in rats after cerebral ischemia, we decided to investigate by immunohistochemistry which cell types were the main source of this MMP-13 after an ischemic insult.
In brain preparations from rats after 90 mins of MCA occlusion, we observed that the main source of MMP-13 in the ischemic tissue were NeuN-positive neurons (Figure 5A), showing a strong nuclear and cytoplasmic signal. Neither glial fibrillary acid protein-positive astrocytes nor Iba-1-positive microglia/inflammatory cells showed any significant staining (Figures 5B and 5C). Some CNPase-positive oligodendrocytes, however, also presented nuclear stain for MMP-13 (Figure 5D).
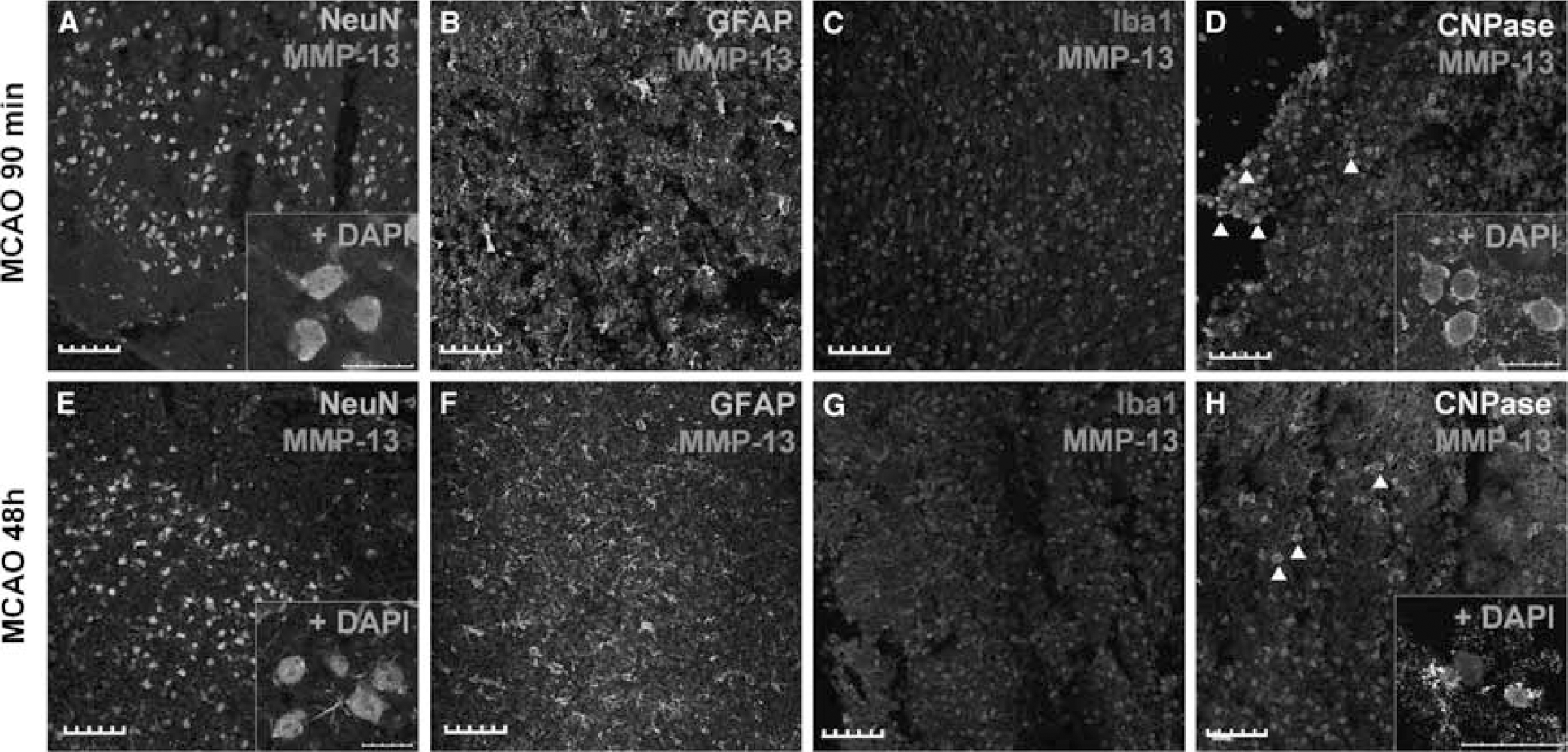
MMP-13 immunohistochemical analysis in rat brains after 90 mins and 48 h of pMCAO. MMP-13 reactivity was basically found in neurons at both time points (
Rat brain preparations from animals subjected to 48 h of ischemia showed a strong nuclear and cytoplasmic MMP-13 immunoreactivity in neurons (Figure 5E), not only in the striatum but also in the ischemic cortex (data not shown). No astrocytes were found to produce MMP-13 at this time (Figure 5F) and few isolated inflammatory cells colocalized with MMP-13 signal (Figure 5G). Nevertheless, some oligodendrocytes also appeared stained for MMP-13 in their nucleus (Figure 5H).
MMP-13 is Found Mainly in Neurons of Humans After Stroke
We also analyzed the subcellular localization of MMP-13 in samples from cortical infarctions and periinfarction regions in human brain samples.
Within the infarction, MMP-13 was found to be mainly produced by NeuN-positive neurons (Figure 6A) and glial fibrillary acid protein-positive astroglia (Figure 6B), in both cases nuclear and cytoplasmic staining was found. Some isolated inflammatory cells were also found positive for MMP-13 (Figure 6C). However, in this case, no CNPase-positive oligodendrocytes were found stained for MMP-13 (Figure 6D).
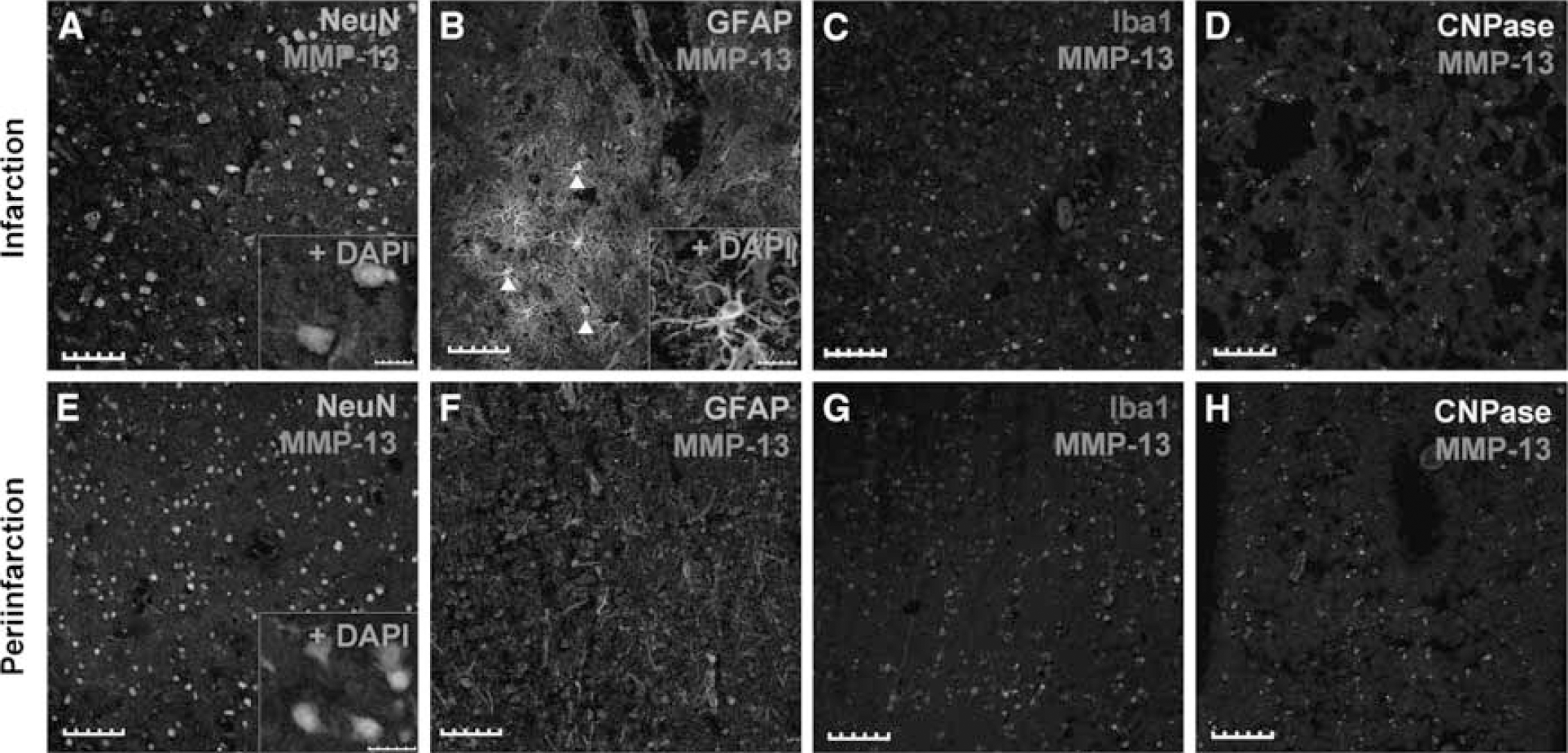
MMP-13 immunohistochemical analysis of human brain samples from the infarct core and the periinfarct area. MMP-13 reactivity was mainly found in neurons in both areas (
In the infarct boundary, MMP-13 was found mainly in neurons (Figure 6E) and no relevant signal was found in astrocytes (Figure 6F), inflammatory cells (Figure 6G), or oligodendrocytes (Figure 6H).
Oxygen and Glucose Deprivation Promotes Neuron Nuclear Translocation of MMP-13 In Vitro
We hypothesized that MMP-13 partially switch its distribution from cytoplasm to cell nucleus after an ischemic stimulus. In an attempt to show this, primary cortical neurons from rats were cultured and subjected to oxygen and glucose deprivation (3 h), followed by a reoxygenation for 24 h. In OGD-treated cells, viability was reduced by 21.55 ± 3.95% measured by MTT reduction (P < 0.001, Figure 7A) compared with controls. Control neurons, kept in the standard incubator with balanced salt solution medium supplemented with 5.5 mmol/L glucose, showed low MMP-13 reactivity restricted to cytoplasm only (Figure 7B). However, cells subjected to OGD, showed high immunoreactivity against MMP-13 in the cell nucleus (Figure 7C). Besides, cytoplasms from hypoxic cells also showed slightly more metalloproteinase signal than cells kept in normoxic conditions. MAP-2, a commonly used neuronal marker, showed intact neurite extensions in normoxic condition, although this staining became discontinuous along the axons of cortical neurons submitted to hypoxia (Figures 7B and 7C).
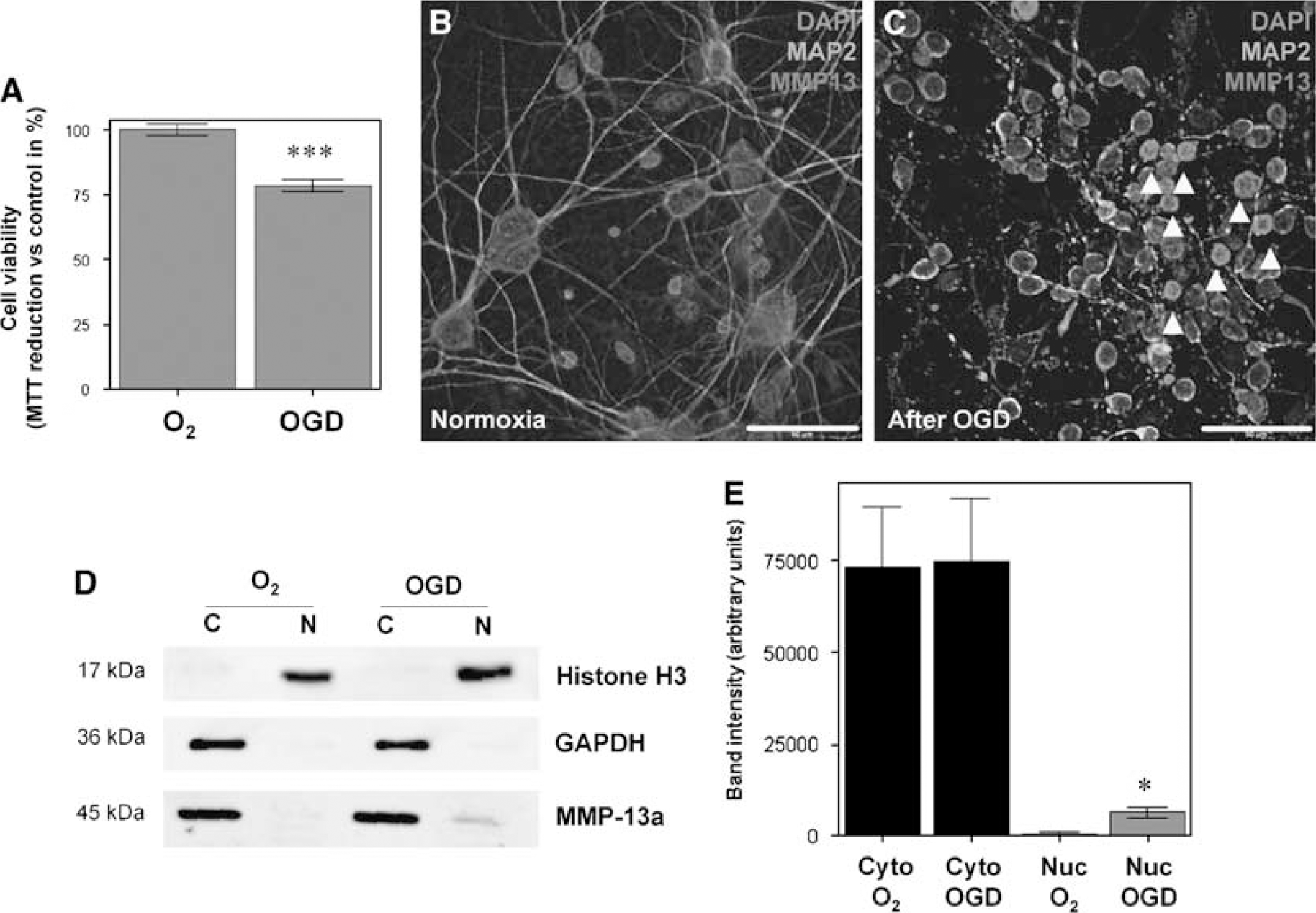
MMP-13 analysis in rat cultured neurons after OGD treatment. (
To semiquantify this nuclear shift, cytosolic and nuclear extracts were obtained. By Western blot, nuclear extracts were found to be uncontaminated by cytosolic fractions, as no positive GAPDH band was found (Figure 7D). Similarly, cytosolic extracts were found to be free of histone H3, a typical nuclear marker. MMP-13 immunoblot showed that while no differences in cytosolic extract were detected, the uclear extract displayed a slight but significant increase of MMP-13 in cells subjected to OGD (P < 0.05, Figures 7D and 7E).
Discussion
In this study, we characterized a part of the MMPs proteolytic activation after ischemic stroke. First, we observe an increased gelatinolytic activity in a rat model of permanent focal ischemia as early as 30 mins after the MCA occlusion. Moreover, we show how this gelatinase activity was first and foremost detected in cell nuclei of brain cells and how, after 2 h of permanent ischemia, began to increase in cytoplasm. Besides, among all MMPs, we report that active MMP-13 is upregulated in rats and humans after stroke, mostly in neurons and oligodendrocytes. Finally, we describe that MMP-13 appears within the cell nuclei in brain tissue after ischemia and we reproduce this effect in vitro in a neuronal culture submitted to oxygen and glucose deprivation.
Our gelatinase in situ results showed an early activation of MMPs after an ischemic insult in rats. The fluorescent substrate used in this assay is efficiently digested by most, if not all, gelatinases and collagenases such as MMP-2, -9, -1, -8, and -13. Previous results from our laboratory revealed a relationship between high plasmatic levels of MMPs-9 and -13 and brain lesion growth in stroke patients (Rosell et al, 2005). Also, we described that MMP-9 is elevated in brain tissue after both hemorrhagic and ischemic stroke in humans (Rosell et al, 2006). For this reason in this study, we aimed to study MMP-13 expression in brain ischemic tissue.
The present study shows that MMP-13 is induced and is activated in rat and, for the first time, in human brain after cerebral ischemia. Previous studies have shown how MMP-13 production is increased after transient cerebral ischemia at delayed times of reperfusion (from 3 to 14 days) in rats (Nagel et al, 2005). Here, we describe that MMP-13 is activated as early as 30 mins of cerebral ischemia in our pMCAO rat model. This is particularly interesting because MMP-13 has a central position in the MMP activation cascade and, for instance, MMPs-2 and -9 are activated by MMP-13 (Leeman et al, 2002). MMPs-9 and -2 activity have been detected in a permanent model of cerebral ischemia at 6 and 24 h after stroke, respectively (Romanic et al, 1998). In a transient focal ischemia rat model, both MMPs-9 and -2 were increased at 4 h post-ischemia (Planas et al, 2001). Likewise, in humans, an increased brain expression of MMP-9 has been described (Rosell et al, 2006). Taken as a whole, these results sketch a temporal profile of MMP activation after an ischemic insult, being MMP-13 one of the first activated MMP based on our results. Contrarily to what we found in the in situ gelatinase assay where the maximum activity is reached at 48h of ischemia in our model, MMP-13 activation peaked 90mins after MCA occlusion. These results suggest that, although MMP-13 may have a role in early times of ischemia, other MMPs might be activated at latter times. Despite all these data, there are still many MMPs that have not been studied at this early time of ischemia and that might also play a role in the MMPs cascade activation.
Previous investigations have reported nuclear gelatinolytic activity in models of transient focal ischemia (Amantea et al, 2007; Gasche et al, 2001) but, to our knowledge, this is the first time that nuclear proteolytic activity is reported in a model of pMCAO at these brief periods of ischemia. It is worth noting that a study performed with a tMCAO mice model showed how apoptotic terminal transferase dUTP nick-end labeling-positive cells had nuclear gelatinolytic activity (Gu et al, 2005). In another study, using a tMCAO rat model, the authors suggested that nuclear neuronal protease activation occurs during reperfusion injury (Amantea et al, 2008). On the basis of our results with the rat model, we suggest that nuclear protease activation may occur after an ischemic insult independently of blood supply restoration, something that may be critical in the human setting as stroke patients remain with artery occlusion for long periods unless spontaneous or therapeutic reperfusion occurs.
Human studies of brain parenchyma from strokes are scarce probably because of the difficulty to obtain these samples. There is a report by Anthony et al (1997) in which they describe the presence of MMPs-2, -3, -7, and -9 in ischemic brain, mainly in inflammatory cuffs around vessels. Our study provides new information as we describe the presence of active MMP-13 in the infarct core and in the periinfarction in human brain after stroke.
A striking result is that MMP-13 was present in the cell nucleus after cerebral ischemia, and that we were able to reproduce this peculiar effect in vitro. To our knowledge, this is the first time that an MMP is described to be within the nuclear compartment in brain tissue. Supporting the presence of nuclear MMPs, other studies have described MMPs within cell nucleus in other organs or tissues, that is, MMP-2 has been found in the cell nucleus of cardiac myocytes (Kwan et al, 2004), MMP-3 has been detected in the nucleus of hepatocytes (Si-Tayeb et al, 2006) and MT1-MMP has been isolated in nuclear fractions of hepatocellular carcinoma cells (Ip et al, 2007). Bearing in mind these results, a question arising is what would be the function of nuclear MMPs after stroke? There are few studies that suggest a role for MMPs in cell death pathways. An important work revealed that MMP inhibition after cerebral ischemia reduced DNA fragmentation by 51%, infarct size by 60%, and increased poly-(ADP-ribose) polymerase, a protein involved in DNA repair, by 84%, even in MMP-9-deficient mice (Copin et al, 2005). These authors suggest that MMPs other than MMP-9 are actively involved in ischemia-induced apoptosis. Furthermore, it has been described that MMP-2 is capable of cleaving poly-(ADP-ribose) polymerase in vitro (Kwan et al, 2004).
Another interesting point is how these MMPs can enter in the nucleus, as they are synthesized in the cytoplasm and normally are secreted or membrane anchored (Nagase and Woessner, 1999). With regard to this issue, Si-Tayeb et al revealed interesting data showing that MMP-3 has a nuclear localization signal (NLS), a tag attached to proteins destined for import to the nucleus and that is recognized by receptors termed karyopherins or importins (Lange et al, 2008). Also, they analyzed other MMPs sequences and found that, besides MMPs-2 and -3, MMPs-1, -8, -10, -13, -14, -16, -17, -19, -20, -23A, and -24 carry some type of putative NLS (Si-Tayeb et al, 2006). Considering all these data, it is conceivable that after ischemia and as part of the apoptotic cascade, DNA fragmentation occurs while poly-(ADP-ribose) polymerase that repair DNA nicks, may be cleaved by MMPs, besides caspase-3, contributing to apoptosis.
In our experiments, we found that MMP-13 is mainly produced in neurons and it is clearly seen in rats and humans. However, we found some differences between our rat model and human samples. Although rat oligodendrocytes showed high reactivity for MMP-13, in humans we were not able to detect them. This could be explained by the different cytoarchitecture of rats and humans and by the different localization of ischemic lesions. Although in rats, neurons and oligodendrocytes are found together especially in striatum (Irvin et al, 2008), in human these cells are more abundantly found in white-matter areas (Segal et al, 2007). Besides, we also found an astrogliosis in human infarction that we did not find in rats at 48 h after ischemia. This glia produces MMP-13 in humans but this effect is less clear in our model. This difference could be explained because brain samples correspond to patients that had suffered longer periods of ischemia enabling glia cicatrization to occur.
Our study presents some limitations such as the fact that rat and human ischemia periods are different; however, because of the difficulty to obtain human samples, we believe that these offers a valuable novel information for MMP-13. Another issue is that we do not know the artery perfusion status at the death for human brain samples. This fact might influence the results as ischemia-reperfusion situations might extent the initial ischemic injury. Likewise, in rat experiments, we did not measure regional cerebral blood flow before killing so we could not guarantee that the artery was occluded at that time. Besides, in human studies we only reach significance when we compare infarct and periinfarct samples together against contralateral ones, we firmly believe that this is because of the variability of patients and the small sample size used. Finally, our work only describes the presence of MMP-13 in nuclei but we do not go into the molecular mechanisms for this nuclear translocation nor the MMP-13 function within cell nucleus. Both issues are interesting enough to be investigated in future studies.
Collectively, the present study shows that MMP-13 is increased in the very acute phase of cerebral ischemia both in human stroke and animal models. Furthermore, MMP-13 appears in nuclei of, mainly but not only, neurons after an ischemic stimulus. These experiments indicate a novel intranuclear role for MMP-13 that should be further studied, together with other potential nuclear-MMPs, to understand their contribution in the pathophysiology of cerebral ischemia.
Footnotes
Acknowledgements
We are grateful to Manolo Quintana for statistical advice and to all neurologists and residents from the Neurovascular Unit and technical staff from the Neuropathology Department who helped in performing this study. EC is supported by a grant from the Fondo de Investigaciones Sanitarias (FIS 05/322). AR is supported by a Beatriu Pinós Fellowship and MBP by Juan de la Cierva Program. LG-B and MH-G are both supported by Post-Doctoral Fellowship from the FIS. Neurovascular Research Laboratory takes part into the Spanish stroke research network RENEVAS.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.