
Abstract
Activated protein C (APC), a serine-protease with anticoagulant, anti-inflammatory, and cytoprotective activities, is neuroprotective and holds potential to treat different neurologic disorders. It is unknown whether APC crosses the blood—brain barrier (BBB) to reach its therapeutic targets in the brain. By using a brain vascular perfusion technique, we show that 125I-labeled plasma-derived mouse APC enters the brain from cerebrovascular circulation by a concentration-dependent mechanism. The permeability surface area product of 125I-APC (0.1 nmol/L) in different forebrain regions ranged from 3.11 to 4.13 μL/min/g brain. This was approximately 80- to 110-fold greater than for 14C-inulin, a simultaneously infused reference tracer. The Km value for APC BBB cortical transport was 1.6±0.2 nmol/L. Recombinant APC variants with reduced anticoagulant activity, 5A-APC and 3K3A-APC, but not protein C, exhibited high affinity for the APC BBB transport system. Blockade of APC-binding site on endothelial protein C receptor (EPCR), but not blockade of its protease-activated receptor-1 (PAR1) catalytic site, inhibited by > 85% APC entry into the brain. APC brain uptake was reduced by 64% in severely deficient EPCR mice, but not in PAR1 null mice. These data suggest that APC and its variants with reduced anticoagulant activity cross the BBB via EPCR-mediated saturable transport.
Introduction
Activated protein C (APC) is an endogenous serine protease with anticoagulant, anti-inflammatory, and cytoprotective activities (Mosnier et al, 2007a). Its anticoagulant activity is mediated by irreversible inactivation of the coagulation cofactors, factor Va and FVIIIa (Griffin et al, 2002). Independent of its anticoagulant activity, APC exerts direct cytoprotective effects, including antiapoptotic and anti-inflammatory alterations in gene-expression profiles that require APC-mediated activation of protease-activated receptor-1 (PARI; Joyce et al, 2001; Riewald et al, 2002; Riewald and Ruf, 2005).
In the central nervous system, activation of the protein C (PC) cellular pathway results in neuroprotection after transient brain ischemia or embolic stroke (Cheng et al, 2003; Zlokovic et al, 2005). APC inhibits neuronal cell death in various in vitro and in vivo models of neuronal toxicity including N-methyl-D-aspartate-induced excitotoxic lesions, tissue-plasminogen activator-induced toxicity or staurosporine-mediated apoptosis (Guo et al, 2004; Liu et al, 2004). In addition, APC prevents p53-mediated apoptosis in brain endothelium (Cheng et al, 2003), protects endothelial barriers from different types of injury (Feistritzer and Riewald, 2005; Finigan et al, 2005; Isermann et al, 2007), and blocks tissue-plasminogen activator-mediated blood—brain barrier (BBB) breakdown after stroke (Cheng et al, 2006). Its effects on the endothelium require binding to endothelial protein C receptor (EPCR) and PARI activation (Riewald et al, 2002; Cheng et al, 2003; Uchiba et al, 2004). A recent study has shown that an APC variant with reduced anticoagulant activity is neuroprotective in a mouse model of multiple sclerosis (Han et al, 2008).
Although, APC and its variants with reduced anticoagulant activity (Mosnier et al, 2004; Mosnier et al, 2007b) hold the potential to treat different neurologic disorders, it is not known whether these glycoproteins with a molecular weight of approximately 60 kDa (Mosnier et al, 2007a) can cross an intact BBB to reach their therapeutic targets within the brain. To address this question, we studied APC transport from cerebrovascular circulation into the brain by using a mouse brain vascular perfusion technique that has been previously used to characterize blood-to-brain transport of several peptides and proteins (LaRue et al, 2004).
Materials and methods
Reagents
Plasma-derived mouse PC and APC were obtained from Innovative Research (Novi, MI). Mouse recombinant APC variants with reduced anticoagulant activity, that is, 3K3A-APC (KKK191-193AAA) and 5A-APC (RR229/230AA and KKK191-193AAA), were produced as reported (Mosnier et al, 2004; Mosnier et al, 2007b). Antibodies that block (RCR-252) and do not block (RCR-92) APC binding to EPCR were gifts from Dr Fukudome (Saga Medical School, Saga, Japan). Antibody against the catalytic site of PARI (H-111) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Animals
C57BL/6 mice, 2- to 3-month-old (Jackson Laboratories, Bar Harbor, ME), severely deficient EPCR mice (Castellino et al, 2002), and PARI null mice (from Dr Shaun Coughlin, University of California) were used in transport studies. Mice were kept under standard housing conditions and feeding schedules until required for the experimental procedures. All studies were performed according to the National Institutes of Health guidelines using approved institutional protocols.
Radiolabeling
Mouse recombinant APC (10 mg) was radiolabeled using 0.6 mCi Na125I by Iodo-Gen (Thermo Scientific, Rockford, IL). Free iodide was removed from radiolabeled APC preparations by using Zeba gel filtration column (Thermo Scientific). The labeled APC had a specific activity of 2 to 3 μCi/μg and was > 99% trichloracetic acid (TCA) precipitable. 125I-APC was stored in small aliquots at −80°C and used within 24 h of labeling. Before brain perfusion (see below), high-performance liquid chromatography was performed to assure use of intact radiolabeled protein in all animal studies.
99mTc-bovine serum albumin was labeled using stannous tartrate as the reducing agent, as reported (Pettit et al, 1980). Briefly, 1 mg bovine serum albumin was labeled with ~ 5 mCi 99mTc-pertechnetate at pH 3.1 to 3.2, in the presence 20 μmol/L stannous tartrate (Sigma, St Louis, MO, USA). Labeled 99mTc-bovine serum albumin was purified by Zeba desalting column (Thermo Scientific). 99mTc-bovine serum albumin > 99% TCA precipitable was used in all animal studies.
Brain Perfusion Technique
To determine transport of 125I-APC at the BBB, we used a mouse brain perfusion method described in detail elsewhere (LaRue et al, 2004). Mice were anesthetized by i.p. injections with a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg). The right common carotid artery was isolated and cannulated with polyethylene tubing (PE10) connected to the perfusion system (LaRue et al, 2004). Mouse brains were perfused at 1.0 ml/min with washed sheep red blood cells suspended in an artificial plasma solution containing (mmol/L): 123 NaCl, 4 KCl, 2.5 CaCl2, 1.8 MgCl2, 25 NaHCO3, 1.2 KH2PO4, 5.5 D-Glucose, and 6% dextran (MW 70,000, Sigma). The final hematocrit was approximately 20%. The perfusate was gassed with 95% O2/5%CO2, warmed to 37°C, and filtered by passing the perfusion solution through polymer wool (Polyester Fiberfill, Jo-Ann Stores, Inc., Hudson, OH) before entering the cerebral circulation. The temperature and the perfusion pressure were continuously monitored. The acid-base status and blood gases were monitored at frequent intervals using an ABL70 acid-base analyzer (Radiometer America, Weshake, OH). During the brain perfusion, the perfusion pressure was kept elevated by approximately 15 mmHg above the animal's arterial blood pressure to prevent any possible inflow from the systemic circulation. At the start of the perfusion, the contralateral common carotid artery was ligated and both jugular veins severed to allow free drainage of the perfusate. The brain was initially perfused with a tracer-free medium for 10 mins to allow for physiologic equilibration and stabilization before experimental procedures. Before entering the cerebral circulation the perfusion medium was mixed with tracer infusate in a perspex block that served as a mixing chamber. The perfusion was terminated by decapitating the animal at predetermined times.
Experimental Design
To determine transport across the BBB from the cerebrovascular circulation into the brain, mouse plasma-derived 125I-APC (0.1 nmol/L) was infused simultaneously with the reference tracers 14C-inulin (extracellular space marker) and 99mTc-albumin (vascular space marker) into the perfusion circuit by a slow-drive syringe pump (Harvard Apparatus, Holliston, MA) at a rate of 0.1 mL/min. The levels of 125I-APC at 0.1 nmol/L in the arterial circulation were close to its physiologic concentrations in mouse plasma as determined by mouse APC-specific enzyme-linked immunosorbent assay (Fernandez et al, 2006). The time dependency of 125I-APC (0.1 nmol/L) uptake into the brain was determined by perfusing the radioactive tracers mixture for various time periods from 1 to 10 mins. The concentration dependency of APC uptake into the brain was studied by perfusing brains during linear phase of 125I-APC (0.1 nmol/L) uptake that was within 10 mins, in the presence of various concentrations of unlabeled APC ranging from 0.5 to 50 nmol/L. The effects of unlabeled mouse APC variants 3K3A-APC and 5A-APC and mouse protein C at 10 nmol/L, on uptake of 125I-APC (0.1 nmol/L) were studied within 10mins. These studies were performed to determine whether APC variants or protein C share with APC a common transport system at the BBB. The effect of antibodies that block (RCR-252) and do not block (RCR-92) APC-binding site on EPCR and an antibody that blocks PAR1 catalytic activation site (H-111; Cheng et al, 2003) on 125I-APC (0.1 nmol/L) BBB uptake were studied at 20 μg/ml within 10 mins. These studies were performed to establish whether APC transport at the BBB requires EPCR or PAR1. In all inhibition experiments, brains were initially perfused for 5 mins with unlabeled potential competitors or inhibitors without tracers and then for 10 mins together with tracers. Finally, 125I-APC (0.1 nmol/L) was infused with control tracers for 10 mins in severely deficient EPCR mice (Castellino et al, 2002) and PAR1 null mice.
Analysis of Radioactivity
The pial vessels and choroid plexuses were removed and the ipsilateral (right) cerebral cortex, caudate nucleus, and hippocampus dissected and homogenized for radioactivity measurements. Perfusion medium was centrifuged and the supernatant was prepared for radioactivity measurements. In all experiments, 99mTc-albumin and 125I-APC in brain and arterial inflow samples were subjected to TCA precipitation and the radioactivities determined in the γ-counter (Wallac Vizard Gamma Counter, PerkinElmer, Meriden, CT). The high-performance liquid chromatography analysis of 125I-APC radioactivity in the arterial inflow supernatant and brain homogenates was additionally performed in separate set of experiments to confirm the results of TCA precipitation analysis. For 14C counting, the samples were solubilized in 0.5 ml tissue solubilizer (PerkinElmer, Boston, MA) overnight, followed by addition of 5 ml of scintillation cocktail (Packard Ultima Gold); samples were analyzed in a liquid scintillation spectrometer (Packard Tri-Carb 2100TR, PerkinElmer, Shelton, CT).
Calculations
125I-APC and 14C-inulin uptake into the cerebral cortex was expressed as distribution volume, Vd (μl/g), as reported (Zlokovic et al, 1989). 125I-APC and 14C-inulin uptake was corrected for the residual vascular radioactivity using 99mTc-albumin, as indicated in Equations 1 and 2.


Equations 1 and 2 were used to determine Vd values for125I-APC and14C-inulin in the caudate nucleus and hippocampus.
The permeability surface area (PS) product was calculated using Equation 3, as reported (Zlokovic et al, 1989).
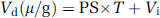
where T is the perfusion time in seconds (s) and Vi the initial volume of distribution. PS values were expressed as μL/min/g brain, as reported (Zlokovic et al, 1989).
The Michaelis—Menten analysis was applied to determine the affinity constant Km and the maximal transport rate Vmax of APC BBB transport system by using Equation 4, as reported (Zlokovic et al, 1989).
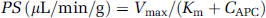
where CAPC is the varying concentration of APC in the circulating perfusion fluid. Kinetic constants were obtained by nonlinear regression curve fitting (Prism 3.0).
Activated protein C influx (Jin) was computed as reported (Zlokovic et al, 1989)
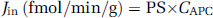
The inhibitory constant (Ki) was determined from cross-inhibition experiments using the velocity ratios, as reported (Zlokovic et al, 1990).
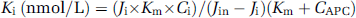
where J in and Ji are 125I-APC influx values in the absence and presence of the inhibitory concentrations of APC variants or protein C, and Ci and CAPC are concentrations in the perfusion fluid of the inhibitory APC variants or protein C and 125I-APC, respectively.
Brain Capillary Depletion
In separate experiments, microvessels were isolated from brain, as described (Wu et al, 2003). To minimize diffusion of 125I-labeled APC out of the capillaries during isolation, all steps were performed at 4°C. Briefly, after perfusion with radiolabeled tracers, animals were decapitated, ipsilateral hemisphere removed, quickly weighed, and cut into small pieces on ice-cold dish at 4°C. These were then homogenized in 3.5-fold excess volume of ice-cold buffer solution at 4°C containing (mmol/L): NaCl (103), KCl (4.7), CaCl2 (2.5), KH2PO4 (1.2), MgSO4 (1.2), HEPES (15), NaHCO3 (25), glucose (10), sodium pyruvate (1), at pH 7.4, in a 5-ml hand-held glass dounce tissue grinder (0.127 mm clearance, Kontes Glass Co., Vineland, NJ) using 8 to 10 up-and-down strokes. The homogenate was suspended in an equal volume of 26% dextran (average molecular weight 64,000 to 76,000, Sigma-Aldrich Inc., St Louis, MO), transferred to a 1.5 ml preweighed Eppendorf tubes and spun at 5800g (Eppendorf 5415R, Hamburg, Germany) for 15 mins at 4°C. The pellet was carefully separated from the supernatant. The radioactivity was determined in the supernatant containing the capillary-depleted brain, homogenized cerebral cortex, vascular pellet, and the perfusate. This method has been used previously to determine whether peptides and proteins enter the capillary-depleted brain (Triguero et al, 1990). The Vd values of APC in capillary-depleted brain and vascular pellet were corrected for inulin uptake as well as for their respective weights per gram of the whole-brain weight. Typically microvascular pellet was <3% of the whole-brain wet weight and the remaining represented capillary-depleted brain.
Western Blot Analysis
Cerebral cortex microvessels were isolated from EPCR+/+ and severely depleted EPCRd/d mice, as described above. Brain microvessels were lysed, and protein samples (40 mg) separated on a 10% SDS-polyacrylamide gel and transferred to nitrocellulose membrane. For detecting EPCR, the membrane was blocked for 1 h in 5% nonfat milk Tris-buffered saline and then probed with a rabbit anti-human polyclonal antibody that cross-reacts with mouse EPCR (1:1000; Invitrogen) at 4°C overnight. After washing with Tris-buffered saline, the membrane was incubated with HRP-conjugated goat-anti-rabbit antibody (1:2000; Dako) for 1 h. The membrane was then washed and developed using an ECL chemiluminescent detection system (GE Healthcare UK Limited, Buckinghamshire, UK).
Statistical Analysis
The results were compared by analysis of variance and Student's t-test, with statistical significance set at P < 0.05.
Results
Figure 1A shows time-dependent uptake of 125I-APC (0.1 nmol/L) TCA-precipitable radioactivity into the cerebral cortex after correction for 99mTc-albumin residual vascular radioactivity. 125I-APC uptake was linear within the 10 mins studied period of time (R2 = 0.9860). After 1 and 10 mins of brain perfusion, the TCA-precipitable fractions of 125I-APC in the cerebral cortex were > 97% and 94%, respectively (n = 3 to 5 brains per time point), compared with approximately 99% found in the arterial inflow, indicating minimal degradation of APC during BBB transport over the studied period of time. The TCA precipitation results showing intact 125I-APC in the arterial inflow and > 94% intact APC in the brain homogenate at the end of 10 mins perfusion experiment were additionally confirmed by high-performance liquid chromatography analysis (not shown). Extremely low 14C-inulin uptake corrected for 99mTc-albumin distribution was also linear with time (R2 = 0.9924).
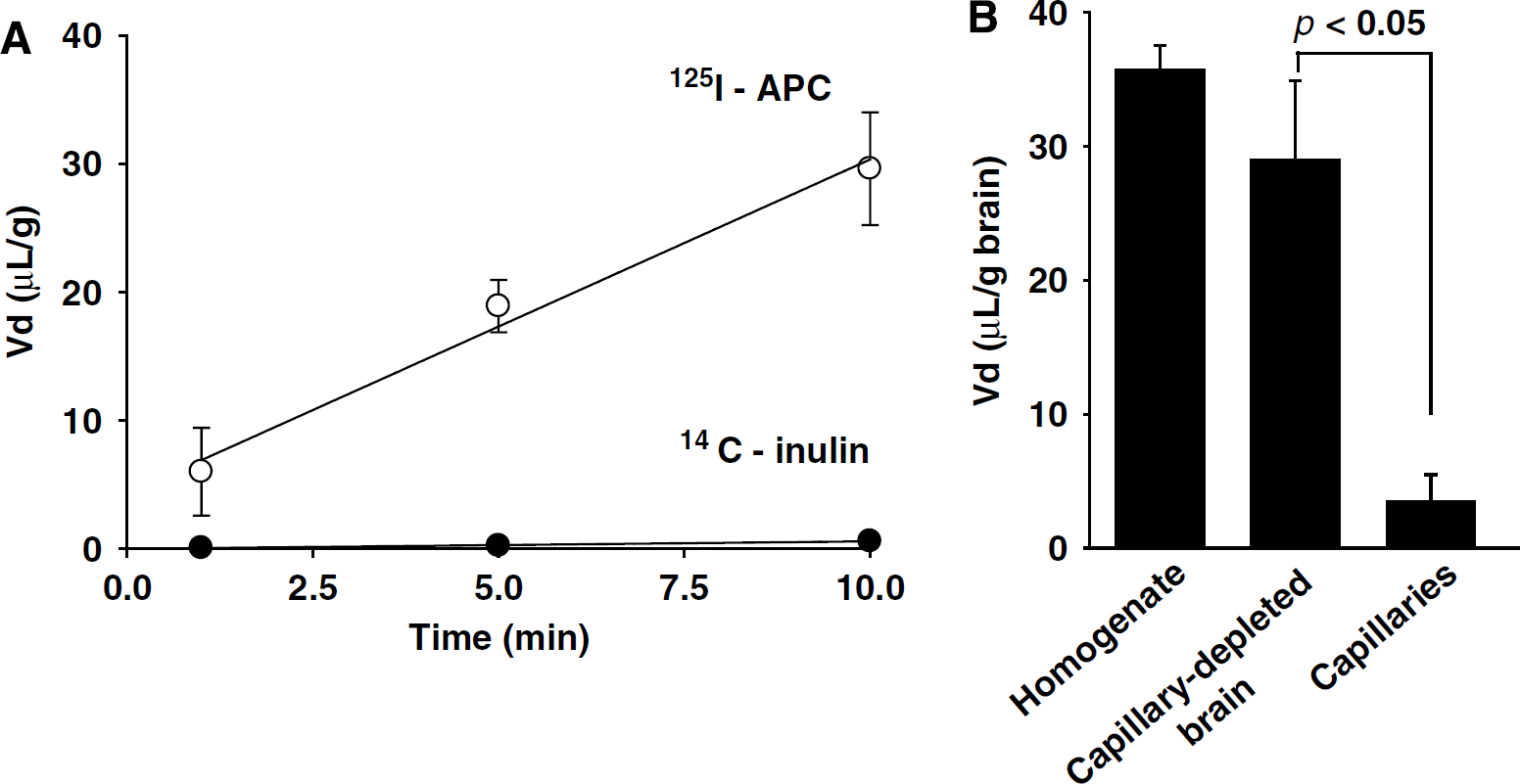
Time-dependent transport of 125I-labeled plasma-derived mouse APC across the mouse blood—brain barrier studied by the vascular brain perfusion method. (
The PS products of 125I-APC and 14C-inulin in the parietal cortex, caudate nucleus, and hippocampus are given in Table 1. Table 1 shows that circulating 125I-APC at a concentration of 0.1 nmol/L, which is close to physiologic plasma APC levels in mice (Fernandez et al, 2006), entered the brain at a rate ranging from 3.1 to 4.1 μL/min/g brain. The rates of 125I-APC entry across the BBB were 78-to 110-fold greater than the corresponding values of simultaneously infused extracellular space reference marker 14C-inulin. The y-intercept of 125I-APC linear uptake line indicated an initial rapid distribution space, Vi (Equation 3), of APC of 4.37±1.09 mL/g that was significantly (P < 0.05) greater than the Vi value for 14C-inulin that was barely different from zero. Previous work has shown that the Vi values significantly higher than zero typically reflect an initial binding of a given peptide or protein to their respective putative carriers or receptors at the luminal side of the BBB (Patlak et al, 1983).
Brain capillary PS products (μL/min/g) of 125I-APC (0.1 nmol/L) and 14C-inulin in the perfused mouse brain

APC, activated protein C.
Values are mean±s.e.m., n = 3 to 5.
The permeability surface area products were calculated using Equation 3.
To confirm that 125I-APC enters the brain, we performed a capillary-depletion experiment. Figure 1B shows that within 10 mins of perfusion the majority of 125I-APC (< 85%) was in the capillary-depleted brain fraction, while only a minor portion remained sequestered to microvessels isolated from brain.
Next, we studied whether APC uptake at the BBB is concentration-dependent.
Figure 2A shows that the PS product of 125I-APC (0.1 nmol/L) was progressively reduced by the increasing levels of unlabeled APC in the perfusion medium from 0.5 nmol/L to 50 nmol/L, indicating the presence of a saturable, carrier-mediated and/or receptor-mediated transport system for APC at the BBB. The analysis of APC influx into cerebral cortex confirmed a concentration-dependent and saturable transport (Figure 2) with an affinity constant, Km, of 1.57±0.14 nmol/L and a maximal transport capacity, Vmax, of 7.2±1.0fmol/min/g brain (Table 2).
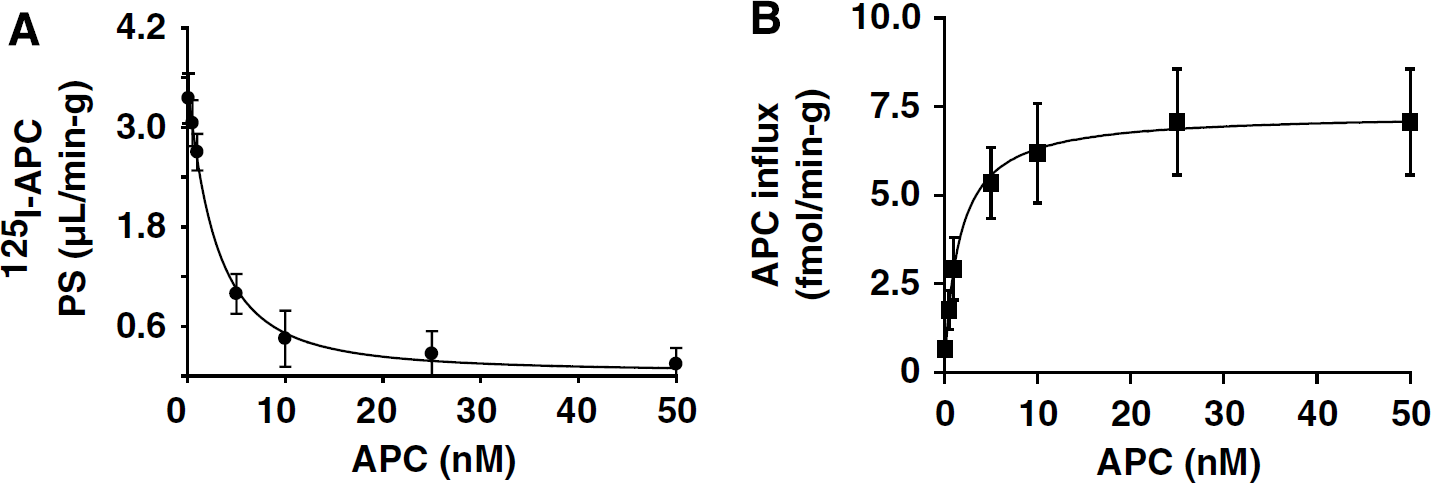
Concentration-dependent transport of 125I-labeled plasma-derived APC into the brain. (
Kinetic parameters of APC uptake in mouse cerebral cortex
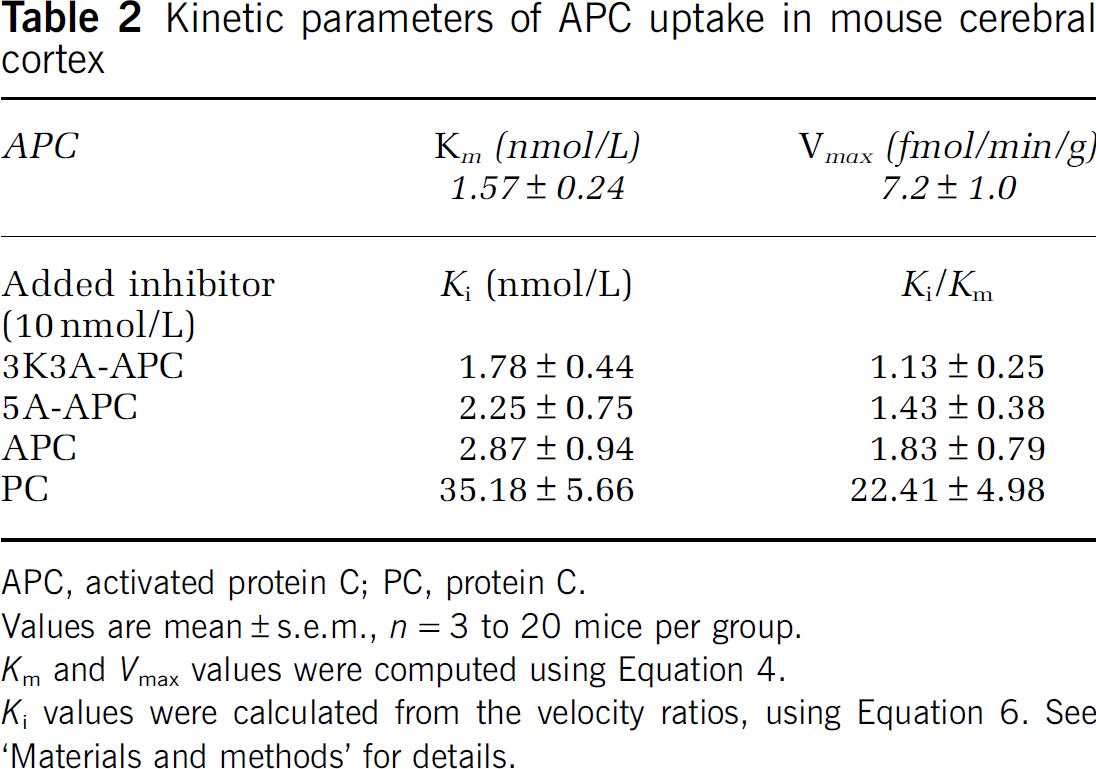
APC, activated protein C; PC, protein C.
Values are mean±s.e.m., n = 3 to 20 mice per group.
Km and Vmax values were computed using Equation 4.
Ki values were calculated from the velocity ratios, using Equation 6. See
'Materials and methods' for details.
APC variants with greatly reduced anticoagulant activity, that is, 3K3A-APC and 5A-APC, at 10 nmol/L almost abolished 125I-APC (0.1 nmol/L) uptake into the cerebral cortex, suggesting a significant cross-inhibition. In contrast, mouse PC at 10 nmol/L had only a modest inhibitory effect (Figure 3). The inhibitory constants, Ki (nmol/L), of 3K3A-APC, 5A-APC, and PC were calculated using Equation 6 and compared with that of plasma-derived APC (Table 2). Although the Ki/Km ratios of APC variants suggested somewhat lower affinity of 3K3A-APC and 5A-APC for the APC transport system at the BBB in vivo compared with plasma-derived APC, these differences did not reach statistical significance. Moreover, protein C zymogen had > 22-fold lower affinity for APC BBB transport system compared with plasma-derived APC. These data indicate that APC variants share with APC the same putative transport system at the BBB to enter the brain, whereas protein C has substantially lower affinity than APC for blood-to-brain transport.
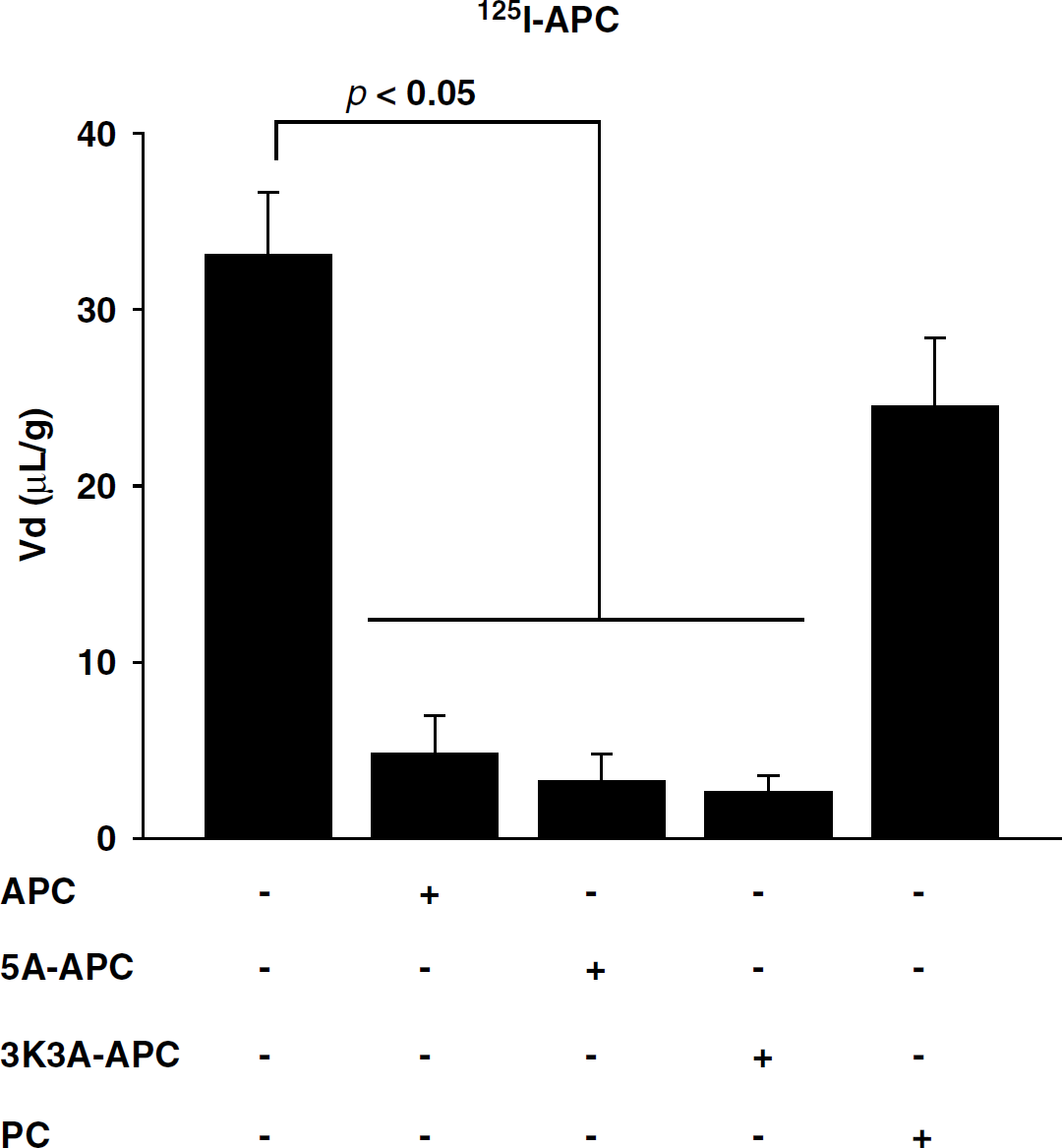
APC variants with reduced anticoagulant activity cross-inhibit 125I-labeled plasma-derived APC transport into the brain. TCA-precipitable 125I-APC (0.1 nmol/L) uptake into the parietal cortex corrected for the vascular space distribution (99mTc-albumin) after 10 mins of cerebrovascular arterial perfusion in the absence and presence of 10 nmol/L of unlabeled APC, 5A-APC, 3K3A-APC, and mouse plasma-derived protein C (PC). Values are mean±s.e.m., n = 3 to 6 mice per group.
We then explored whether APC brain endothelial receptors EPCR and PAR1 (Cheng et al, 2003; Thiyagarajan et al, 2007) are required for APC transport into the brain. An antibody that blocks specifically APC-binding site on EPCR (RCR-252) inhibited uptake of 125I-APC into the cerebral cortex by 85.2%, whereas a control EPCR antibody (RCR-92) that does not block APC-binding site on EPCR did not have any effect on APC transport (Figure 4A). An antibody that blocks the activation of PAR1 (H-111) had no effect on 125I-APC uptake into the brain (Figure 4A). To confirm that EPCR is required for BBB transport of APC and that PAR1 is not needed for the transport process, we next studied brain uptake of APC in severely deficient EPCR mice and PAR1 null mice. The TCA-precipitable 125IAPC (0.1 nmol/L) uptake into cerebral cortex was reduced by approximately 64±4% in mice severely deficient in EPCR (Figure 4B). These mice express substantially decreased levels of EPCR in brain microvessels than control mice (< 15% of control values) as shown by Western blot analysis of isolated microvessels from EPCR+/+ and EPCRδ/δ mice (Figure 4C). In contrast, 125I-APC brain uptake was unchanged in PAR1 null mice compared with control mice (Figure 4B).
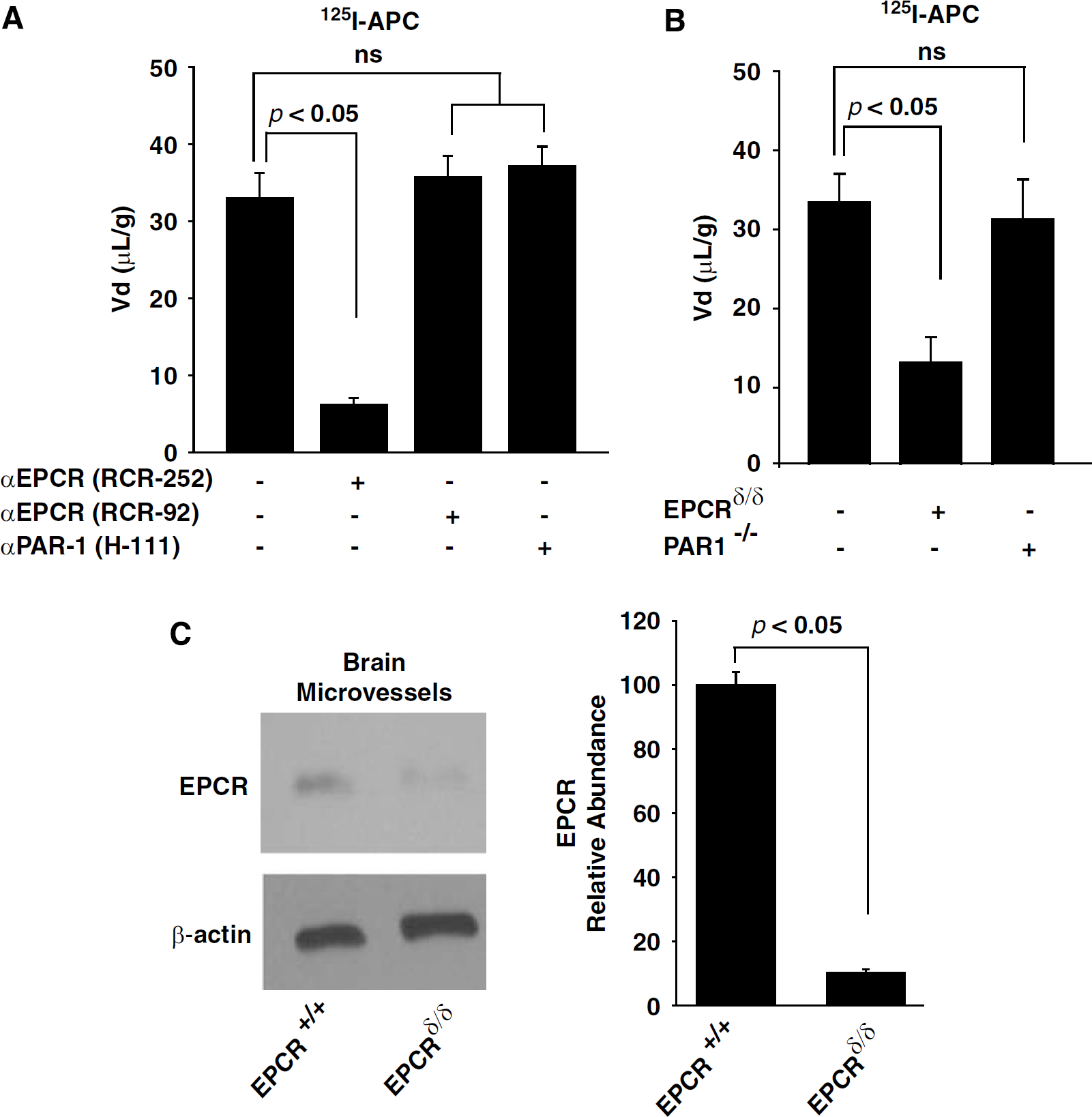
APC transport across the BBB from the cerebrovascular circulation into the brain requires EPCR. (
Discussion
The present study provides first direct evidence for a concentration-dependent EPCR-assisted transport of circulating APC into the brain in vivo across an intact BBB. The regional BBB PS product for 125I-APC at APC's cerebral arterial concentration comparable to physiologic plasma levels of endogenous mouse APC (Fernandez et al, 2006) ranged from 3.1 to 4.1 μL/ mins/g brain or two orders of magnitudes greater than for the extracellular space reference marker inulin. This suggests a significant unidirectional transport of APC from cerebral arterial blood to brain under normal conditions. The kinetic analysis revealed that the transport system for APC has a high affinity, that is, Km = 1.6 nmol/L, but relatively low capacity (Vmax = 7.2 fmol/mins/g brain). As plasma APC concentration in mice is close to 0.1 nmol/L, the BBB transport system with a Km value that is approximately 15-fold higher would favor a continuous delivery of small amounts of APC from plasma to brain at any time. This in turn could be of physiologic importance for APC's effects in the CNS such as immunologic surveillance and/or cerebral protection, because APC's precursor protein C is not synthesized normally in the CNS (Jamison et al, 1995) or is expressed at very low levels, that is, < 1% of those found in the liver (Yamamoto and Loskutoff, 1998).
Although it is established that the BBB restricts the uptake of hydrophobic molecules into the brain, several specific carrier-mediated transport systems for essential nutrients such as glucose and amino acids have been described at the luminal surface of the BBB, as well as specific receptor-mediated transport systems for different proteins and peptides (Zlokovic, 2008). In contrast to rapidly transporting nutrients, studies of slowly penetrating peptides and proteins across the BBB require approaches that would allow for an extended exposure time of studied test-molecules to the BBB, that is, from a few seconds to 10 mins or more. This has been achieved by a brain perfusion technique (Takasato et al, 1984; Zlokovic et al, 1986) that has been adapted to transgenic mouse models (LaRue et al, 2004; Banks, 2006).
The PS product of APC obtained in the present study was comparable to PS products reported for peptides such as leucine enkephalin (Zlokovic et al, 1989) and arginine vasopressin (Zlokovic et al, 1990), using brain perfusion method, but was lower than for receptor-mediated BBB transport of amyloid β-peptide-40 (Martel et al, 1996) or apolipoprotein J (Zlokovic, 1996) by 1.4- to 1.9-fold, respectively. Moreover, the PS product of APC was greater than the PS BBB values for insulin (Duffy and Pardridge, 1987), apolipoprotein E4 (Martel et al, 1997), or immunoglobulin G (Zlokovic, 2008), by three to five fold, respectively. Therefore, the rate of APC transport into the brain falls within the middle range of values typically found for different peptides and proteins transport at the BBB.
Our study shows that two APC variants with reduced anticoagulant activity, that is, 3K3A-APC and 5A-APC, cross-inhibit APC BBB transport and have comparable inhibitory constants (Ki) for the APC transport system at the BBB as plasma-derived APC. Although, the Ki/Km ratios suggested somewhat lower affinity of 3K3A-APC and 5A-APC for BBB transport than of APC itself, these differences were not significant. The anticoagulant action of APC involves a cleavage site at Arg506 in factor Va that depends on positively charged residues in surface loops on APC's protease domain including loop 37 (residues 190 to 193), the Ca2+ -binding loop (residues 225 to 235), and the autolysis loop (residues 301 to 316) (Mosnier et al, 2007a). The two APC variants were generated with alanine mutation in the 37 loop, that is, 3K3A-APC (KKK191-193AAA) (Mosnier et al, 2004), and in the 37 and Ca2+ -binding loops, that is, 5A-APC containing 5 alanine substitutions for five protease domain Arg229/230 and Lys 191 to 193 residues (Mosnier et al, 2007b). These APC mutants exhibited little or almost no anticoagulant activity (< 5%), but retained normal antiapoptotic activity that required PAR1 and EPCR on endothelial cells (Mosnier et al, 2004; Mosnier et al, 2007b). As the Gla-domain of APC interacts with EPCR (Mosnier et al, 2007a), one would expect that mutations in the exosite loops in APC for interactions with factor Va will not alter significantly the ability of APC to interact with EPCR at the BBB, which we show resulted in transport of circulating APC into the brain.
Endothelial protein C receptor is expressed on endothelial cells where it binds PC and APC specifically, selectively, and saturably (Fukudome and Esmon, 1994). APC and PC bind to EPCR on the surface of isolated human umbilical vein cells with similar affinity (Fukudome and Esmon, 1994), although soluble EPCR inhibits with somewhat higher affinity binding of APC compared with PC to phospholipid vesicles (Liaw et al, 2000). PC binding to soluble EPCR and phospholipids is broadly dependent on correct Gla domain folding and can be influenced by Gla domain mutations (Preston et al, 2005). When thrombin binds to thrombomodulin on endothelial cell surface, its potent procoagulant functions are reversed, and its substrate specificity is directed to PC, which it activates (Esmon, 2003). EPCR augments PC activation by 20-fold in vivoby binding PC, which concentrates PC on the endothelial surface reducing the Km for PC activation by thrombin-thrombomodulin complex (Fukudome et al, 1998).
In the present study, we found that APC transport at the BBB requires EPCR. This has been confirmed both by using specific blocking antibodies for APC-binding site on EPCR and severely deficient EPCR mice with appropriate controls. In contrast to APC, PC had significantly lower affinity for this BBB transport mechanism, that is, by > 22-fold. It is possible that APC that is normally generated from PC on the luminal side of the BBB is rapidly endocytosed after its activation by EPCR and transported across the BBB, in addition to exerting anticoagulant activity in cerebral microcirculation and/or cytoprotective activity via PAR1 (Cheng et al, 2003). We showed that PAR1 was not involved in APC transport, but this does not rule out a possibility that some other yet to be identified coreceptors and/or intracellular mechanisms interact with EPCR to direct APC transport across the BBB and to keep PC on the endothelial surface. EPCR-mediated internalization of APC and diffusion into the nucleus has been shown in hypoxic brain endothelial cells in vitro (Thiyagarajan et al, 2007), which in turn might influence directly gene expression (Joyce et al, 2001; Riewald et al, 2002; Riewald and Ruf, 2005). As brain does not have thrombin and thrombomodulin to activate PC, it is possible that preferential transport of APC across the BBB might represent an important source of brain APC.
In summary, our findings suggest an efficient EPCR-assisted transport of APC into the brain via a mechanism that is shared with its variants with reduced anticoagulant but normal cytoprotective activity. These findings support development of therapeutic interventions with APC and/or its analogues with reduced anticoagulant activity for different neurologic disorders that might benefit from APC's cytoprotective, neuroprotective, and anti-inflammatory activities.
Disclosure/conflict of interest
Dr. Zlokovic is a scientific founder of Socratech LLC, a start-up biotechnology company with a mission to develop new therapies for the diseases of the aging brain including Alzheimer's and stroke.