
Abstract
A foetal cat exhibiting multiple congenital malformations and meeting the criteria for being considered as a case of true schistosoma reflexum (SR) is described. SR in animals is briefly compared with relatively similar malformation entities in humans. Murine gene mutations producing severe ventral body wall defects associated with anomalies of internal organs and other structures are briefly reviewed. New insights into aetiopathogenic mechanisms possibly implicated in the development of SR are suggested. This is probably the first case of true SR reported in the cat.
A 1-year-old short-haired primigravida queen was admitted to a private veterinary clinic. Following a normal pregnancy, dystocia occurred after the birth of a normal kitten. Radiography revealed the presence of a malformed and two more apparently normal kittens. A caesarean section was performed and the three kittens were removed. Of these, one was alive, and two were stillborn, one of the latter appearing severely malformed. Ovariohysterectomy was then performed. The previous clinical history of the dam revealed the absence of drug treatments before and during pregnancy. Exposition to environmental toxins or non-prescription products was unlikely in this case.
The affected male kitten weighed 62.5 g (mean weight of the other three members of the litter: 120 g) and showed multiple congenital anomalies which were compatible with schistosoma reflexum (SR). Except for the manubrium and first sternebra, the sternum was cleft, and only the two first pairs of ribs articulated with a very short sternum. The rest of the ribs and costal cartilages were directed dorsolaterally, their internal thoracic surfaces being orientated exteriorly. Only remnants of an incomplete diaphragm were observed. The large fissure of the ventral body wall continued at the levels of the abdomen and pelvis, and terminated just cranial to the genital tubercle. Non-union of the pelvic symphysis was present. The abdominal wall was reduced to a narrow band of skin reflected dorsally. The amniotic membrane was attached at its origin to the skin margin of the thoracoabdominal fissure. In this situation, progressive closure of the umbilical ring and the development of the definitive umbilical cord could not occur because the formation of a free umbilical cord depends embryologically on the normal closure of the thoracoabdominal wall. In our kitten, the vascular elements (umbilical vein and arteries) and the allantoic pedicle, which are normally integrated to form a well delimited cylindrical structure (the umbilical cord) when they are completely enveloped by the amnios, were partially covered by the amniotic membrane being only adhered to it on one side. The umbilical elements entered separately into the abnormal abdominal cavity and attached to the left caudolateral margin of the body wall defect. For these reasons, the thoracic and abdominal organs were completely exposed in a large persistent extraembryonic coelomic cavity, being only covered externally by the chorionic membrane (Fig 1). The heart showed an abnormal orientation. It was rotated 90° counter-clockwise about its craniocaudal axis and appeared transversally located. The ductus arteriosus was still patent. The lungs were hypoplastic and dorsoventrally flattened; however, their lobation pattern appeared normal. The topographical relationships of the heart to the lungs were completely abnormal, so that the heart was only in contact with the cranial border of the lungs. The oesophagus and trachea were collapsed. The stomach was elongated and transversally located. Instead of showing its normal fixed position by means of a narrow mesoduodenum, the duodenum was attached by a large mesentery, and had abnormal course and location. The jejunum was shorter than normal and consisted of a small number of loops. The descending colon was distended with meconium, and ended blindly. The rectum and anal canal were absent. The anus was imperforate. The liver was abnormal in shape, lobation, location and relationships. It formed a quadrangular mass where the fissures dividing the organ into lobes were completely indistinguishable. It was displaced to the left side and in contact with the internal aspect of the left thoracic wall. The kidneys were present and their gross structure appeared normal. However, the left kidney was rotated 180° about its long axis so that its medial (hileal) border faced in lateral direction and was abnormally in contact with the caudolateral angle of the liver. The testes were located cranially to the urinary bladder, flanking bilaterally the caudal segment of the descending colon.
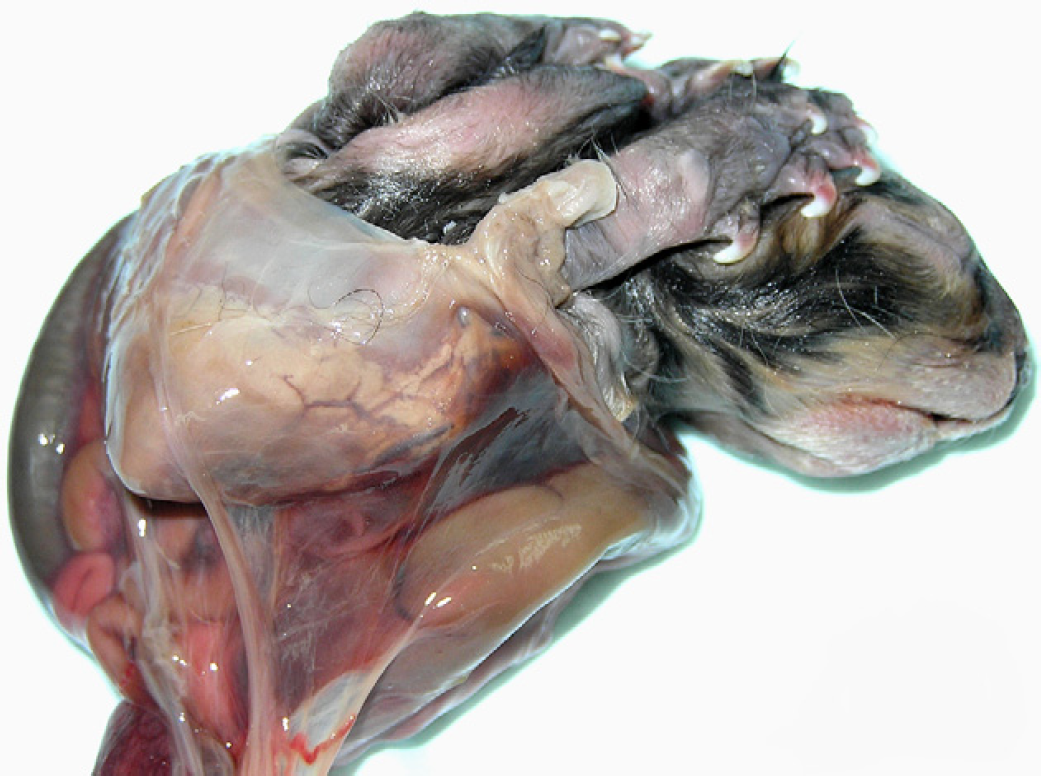
External appearance of the kitten, exhibiting a large thoracoabdominal body wall defect with exposed internal organs enveloped by extraembryonic membranes. Note abnormal positioning of the limbs extended adjacent to the head.
The vertebral column exhibited a series of abnormal and complex curvatures, which could not be returned to normal by gentle manipulation and brought the caudal region in contact with the back. Kyphosis at the middle segment of the cervical column and a pronounced cranial thoracic curvature of ventral convexity (lordosis) were followed by a slight left (lateral) deviation of the middle and caudal thoracic vertebrae (scoliosis). At the thoracolumbar junction, the vertebral column changed abruptly in direction and deviated markedly to the right (scoliosis). Then, the middle and caudal lumbar and the sacral vertebrae directed dorsocranially (lordosis) (Fig 2). A certain degree of vertebral rotation and deformity accompanied the curvature anomalies. A curly tail was present. The four limbs were arthrogrypotic, dorsally directed, and extended adjacent to the head. The forelimbs were abnormally rotated so that the medial aspect of the antebrachii was directed cranially and the palmar aspect of the paws faced laterally (Fig 1). In the head, skull flattening, mandibular brachignathism, slight median cheiloschisis, complete secondary palatoschisis, reduced ossification of the cranial vault bones, and unfused eyelids were observed.

Lateral radiograph showing flattening of the skull, mandibular brachygnathia, and pronounced dorsal spinal flexion (lordosis) at the lumbar vertebral segment.
Ventral body wall closure defects (BWCDs) occur in a wide variety of forms, ranging from uncomplicated hernia into the umbilical cord, omphalocele, and gastroschisis to more complex malformations, such as thoraco- and/or abdominoschisis with diverse degrees of severity, and SR, which can be considered to be the most extreme form. BWCD may occur as isolated anomalies; however, they are often part of a larger constellation of malformations being then included in more complex entities.
SR has long been considered to be primarily a ruminant anomaly (Bezek and Frazer 1994). Cases resembling SR have been reported in other domestic animals, including cats (Kawata and Tiba 1961, Saperstein et al 1976), but the condition has not been recognised in these species because they lacked some particular feature included in the definition based on the characteristic foetal morphology of true SR (Bezek and Frazer 1994, Laughton et al 2005). True SR has recently been more precisely characterised in ruminants as an entity including a series of consistent malformations (abdominoschisis with exposure of abdominal organs, extreme dorsal spinal flexion, limb arthrogryposis, positioning of the limbs adjacent to the head, and lung and diaphragm hypoplasia), and variable defects (thoracoschisis with exposure of thoracic organs, scoliosis, and digestive and/or urogenital system anomalies) (Laughton et al 2005). The cat described here exhibited a spectrum of malformations accurately meeting the criteria for being considered to be a true SR. This is possibly the first case of true SR reported in this species.
A survey of the medical literature for entities consisting of relatively similar phenotypes to those observed in SR animals disclosed mainly three human malformation syndromes. The ‘pentalogy of Cantrell’ (PC) is a heterogeneous entity primarily including sternal, diaphragmatic, cardiac and abdominal wall defects, and exposure of thoracoabdominal organs (Cantrell et al 1958). A variety of additional anomalies (severe scoliosis, cleft lip and/or palate, hypo/aplasia of internal and external genital organs, anal, rectal or other segmental intestinal atresias, and limb defects), extending the originally described PC phenotype, can also be present (Kousseff et al 1996). Most PC cases are sporadic but other cases have been associated with chromosome abnormalities, an X-linked Mendelian mutation, autosomal gene involvement or teratogenic agents (Kousseff et al 1996). The familial occurrence of a combination of defects of the thoracoabdominal wall, diaphragm and thoracic organs, referred to as ‘thoracoabdominal syndrome’ (TAS), was reported by Carmi et al (1990). The TAS gene was later localised to the X chromosome (Parvari et al 1994). ‘Limb–body wall complex’ (LBWC) was established as a descriptive term to categorise a heterogeneous combination of malformations presenting with severe BWCD associated with pronounced kypho- or lordoscoliosis, limb defects, and anomalies of internal organs (Van Allen et al 1987, Russo and Vecchione 1996). The aetiopathogenesis of LBWC remains unclear. Teratogen exposure and a possible genetic origin have been suggested (Martínez-Frías 1997). Based on descriptive observations, divergent theories concerning the pathogenesis of LBWC have been proposed (Brewer and Williams 2004); some of them can explain the external malformations observed in LBWC, but, in our view, they cannot justify the anomalies of internal organs which also occur in this condition.
The precise aetiology and the pathogenesis of SR are unknown. Apparently, no environmental teratogenic agents have been demonstrated to be specifically associated with the development of the condition (Bezek and Frazer 1994). Although the hypothesis of a genetic cause has long been controversial, there are some indications supporting this view. Clusters of cases, in which the same bull had sired affected calves, have been reported, and an autosomal recessive form of inheritance has been suggested (Hámori 1983, Laughton et al 2005).
In mutant mouse studies, more than 30 genes have been implicated in the processes of body wall formation and closure. In most cases, knockout mice exhibited only body wall defects although of varying degrees of severity (Brewer and Williams 2004). However, in certain cases, targeted mutagenesis produced a much wider range of associated anomalies. In particular, two gene families, the Tgfβ signalling molecules and the AP-2 family of transcription factors, appear to be associated with severe forms of BWCD and multiple accompanying anomalies. In Tgfβ2 null mice, the latter included cardiac, lung, cranial (reduced ossification of cranial vault bones), facial (brachygnathia and cleft secondary palate), limb (malrotation and malposition), spinal, sternum, rib, eye, inner ear, and urogenital defects (renal agenesis, renal hypoplasia, hydronephrosis, cryptorchidism, testicular hypoplasia, and uterine horn ectopia) (Sanford et al 1997). The phenotype of Tgfβ2Tgfβ3 double null mice coincided to a large extent with the aforementioned malformations; however, severe thoracoabdominoschisis (lack of distal parts of ribs, lack of sternal primordia, and drastically reduced abdominal wall) with exposure of internal organs was also present (Dünker and Krieglstein 2002). Disruption in mice of the AP-2 gene revealed a wide spectrum of morphogenetic pathways that were variously affected: formation of the neural tube, face, eye, ventral body wall, limbs, spine, heart outflow tract, and urogenital organs. When combined, the interaction of these apparently independent defects resulted in a severe phenotype exhibiting cranial vault bone defects, cheilo/palatoschisis, open eyelids, microphthalmia, thoracoabdominoschisis with exposure of internal organs, limb defects, severe scoliosis, cardiac defects, and urogenital anomalies (Nottoli et al 1998). Interestingly, the heterogeneous spectrum of malformations detected in LBWC infants (Russo and Vecchione 1996) coincided to a large extent with the malformation pattern reported for AP-2 mutant mice. Many of these malformations are also present in SR animals, as well as in our kitten.
Complex malformation patterns including severe BWCD and a wide variety of associated anomalies appear to be aetiologically heterogeneous. In fact, genetic and/or environmental causes can produce relatively similar malformation patterns in humans. It is possible that this could also be the case in SR animals. On the other hand, the great majority of the reported defects are linked to dysmorphogenetic events occurring during early embryogenesis, when the processes of pattern formation in the primary developmental (morphogenetic) field (the bilaminar and then trilaminar embryonic disk) are initiated during gastrulation and early organogenesis. Interestingly, in both Tgfβ2 and AP-2 mutant mice models, the developmental processes most commonly involved in the affected tissues include epithelial–mesenchymal interaction or transition, cell growth and apoptosis, differentiation, extracellular matrix production, cell migration, and tissue remodelling (Sanford et al 1997, Nottoli et al 1998). In our opinion, genetic and/or environmental disruption of these developmental processes could be considered to be an integral part of the aetiopathogenic mechanisms producing these complex malformation patterns including SR.
Footnotes
Acknowledgement
We are greatly indebted to our colleague Mr Guillermo Perna (Clínica Veterinaria ‘La Pedriza’, Manzanares el Real, Madrid, Spain) for providing the malformed cat for study.