
Abstract
A 7-month-old, male European cat was examined because of weakness and inappetence. The cat was dehydrated, polypnoeic and severely weak. Severe, generalised muscle atrophy was present. Spinal reflexes were all decreased to absent. Blood analysis and urinalysis showed several abnormalities, including intermittent hyperoxaluria. The l-gliceric acid concentration was remarkably increased. Electrodiagnostic tests of the peripheral nervous system were abnormal. At necropsy, generalised muscle atrophy was observed. Microscopically, both kidneys showed intraluminal birefringent oxalate crystals. Motor neuron degeneration and accumulation of neurofilaments were observed in the axons of the spinal motor neurons.
Peripheral nerve disorders have been described in cats (Brown 1981, Kramek et al 1984, Lane and deLahunta 1984, Shores et al 1987, Duncan 1991, Braund 2000). Traumatic, metabolic, neoplastic, paraneoplastic, degenerative or inflammatory aetiologies are recognised. Some of the metabolic diseases may be congenital, including feline Niemann–Pick disease (Cuddon et al 1989), gangliosidosis (Braund 2000), glycogenosis (Coates et al 1996), mucolipidosis (Hubler et al 1996), primary hyperchylomicronaemia (Jones et al 1986) and primary hyperoxaluria (McKerrel et al 1989).
Primary Hyperoxaluria (PH) has been described in humans (Marangella et al 1995, Petrarulo et al 1998), dogs (Jansen and Arnesen 1990, Sewell et al 1997) and cats. It was first described in cats in England in 1988 (Blakemore et al 1988, Danpure et al 1989, McKerrel et al 1989). Affected cats lived in a colony, were all relatives and a recessive mode of inheritance could be demonstrated. To our knowledge, no other reports of this disease in cats followed the first description.
In this paper, we describe the physical, neurological, ancillary, histological and ultrastructural examinations of a cat diagnosed to have primary hyperoxaluria, emphasising the differences from the original report.
A 7-month-old, 2.4 kg, sexually intact, male European cat was examined with a 4-week history of weakness and inappetence. The cat was found about 5 months before the onset of the clinical signs; therefore, neither parents nor littermates are known.
On physical examination a moderate dehydration (8%) was detected by testing the skin of the back. Mild polypnoea (respiratory rate: 20/min) was present at rest.
During the neurological examination, the cat was alert and responded normally to visual and auditory stimuli. He was severely weak and reluctant to perform any kind of movement. When forced to move, he could only walk a few steps, demonstrating a plantigrade stance, before lying down. A severe, generalised atrophy affected the muscles of the entire body. Proprioceptive positioning was normal in all four limbs. Other postural reactions could not be evaluated because of the severe weakness. Spinal reflexes were all decreased to absent, whereas cranial nerves and reflexes were normal. Vocalisation became progressively impaired. The lesion was localised to the peripheral nervous system and metabolic, congenital or inflammatory disease was suspected.
The red blood cell (RBC) count revealed a polycythaemia (RBC 12.4×106 – reference range 5.30–8.90). The haematocrit value was 49.6% (reference range 32–48). White blood cell (WBC) and platelets counts were normal. The biochemical profile was normal, except for increased blood urea nitrogen (BUN; 17.85 mmol/l urea – reference range 7.15–11.40 mmol/l urea), creatinine (141.4 μmol/l – reference range 53–125 μmol/l), and lactatedehydrogenase (LDH; 1483 IU/l – reference range 36–253) levels. Feline immunodeficiency virus and feline leukaemia virus tests (Snap Combo Plus; Idexx) were negative as were Toxoplasma gondii antibody titres. A urine sample collected by cystocentesis was cloudy in appearance, had a specific gravity of 1028, and moderate proteinuria. Sediment examination revealed haematuria with 40 RBC per high-powered (400×) microscopic field (reference range 0–10), pyuria with 20 leukocytes per high-powered field (reference range 0–5), cylindruria with five granular casts per low-powered (100×) field (reference range 0–1), and crystalluria with calcium oxalate monohydrate crystals too numerous to count (Fig 1). Bacterial culture was negative. Thoracic radiographs revealed no abnormalities, whereas enlarged kidneys (three times the second lumbar vertebrae on the ventral–dorsal projection) were present on the abdominal radiographs.

Urinary sediment. Several calcium oxalate monohydrate crystals under strong polarised light (unstained, dark field and polarised light ×400).
In the following days, two further urine samples were collected. The findings were similar to those found in the first, but oxaluria was not detected in one (intermittent hyperoxaluria). Because no other causes of oxaluria were likely, primary hyperoxaluria was suspected and specific tests (Blakemore et al 1988) were carried out.
l-Glyceric acid was 32000 μmol while, in a previous study, glycerate was below the limit of detection in the urine obtained from 51 unaffected cats at any age (McKerrel et al 1989).
Electrodiagnostic testing was performed on the cat after premedication with IV diazepam (Valium; Roche) at a dose of 0.5 mg/kg, induction of anaesthesia with IV propofol (Rapinovet; Schering Plough), at a dose of 4 mg/kg, tracheal intubation and maintenance with halothane (Halothane; Merial Italia). On electromyographical examination, fibrillation potentials and positive sharp waves were recorded in the appendicular, paraspinal, and head muscles. The motor nerve conduction velocity of the peroneal-sciatic nerve was moderately decreased (56.8 m/s – reference range 75.2–96.9 m/s). Repetitive stimulation at 3 Hz of the same nerve showed no decremental response. Sensory nerve conduction velocity was not performed.
The final diagnosis was primary hyperoxaluria (l-glyceric aciduria). Because of the worsening of the clinical signs and poor prognosis, the cat was euthanased.
At necropsy, generalised muscle atrophy was observed. The kidneys were enlarged, irregular, pale and firm. Samples from the kidney, liver, spleen, lumbar spinal cord, sciatic nerve and skeletal muscle were removed and fixed in 10% neutral buffered formalin; samples of nervous tissue were fixed in gluteraldehyde 2.5% for ultrastructural studies. Sections were cut at 4 μm thickness and stained with haematoxylin and eosin (HE) and Bielschowsky stain. Immunohistochemical studies were performed using a monoclonal mouse anti-human neurofilament antibody (NF-200) (1:800) (Dako). A specific biotinylated IgG antibody (1:100) (Dako) was used as secondary antibody and 3′3-diaminobenzidine tetrahydrochloride-hydrogen peroxide as chromogen.
Microscopically, large amounts of oxalate crystals were present in both kidneys, which were better appreciated under polarised light, occupying mainly the lumen of the tubules but also the interstitium (Figs 2 and 3). Other lesions: glomerulosclerosis, multifocal non-purulent interstitial nephritis, and interstitial fibrosis were present. Neuropathological findings affected the lumbar spinal cord and peripheral nerves. In the spinal cord, several motor neurons were enlarged and degenerated. Large swellings along the proximal length of the axons of the spinal motor neurons were seen especially with Bielschowsky stain. The sciatic nerve also demonstrated enlarged axons and Wallerian degeneration. Immunohistochemistry for 200 kDa neurofilament confirmed that enlargement of the perikaria and the axons were due to accumulation of phosphorylated neurofilaments. Ultrastructural studies confirmed the accumulation of abnormal neurofilaments in affected neurons. Oxalate crystals within the nervous tissue were not observed. Finally, skeletal muscles showed variation in fibre size, showing some fibres clearly atrophic while others were hypertrophic, and some of them contained internal nuclei.
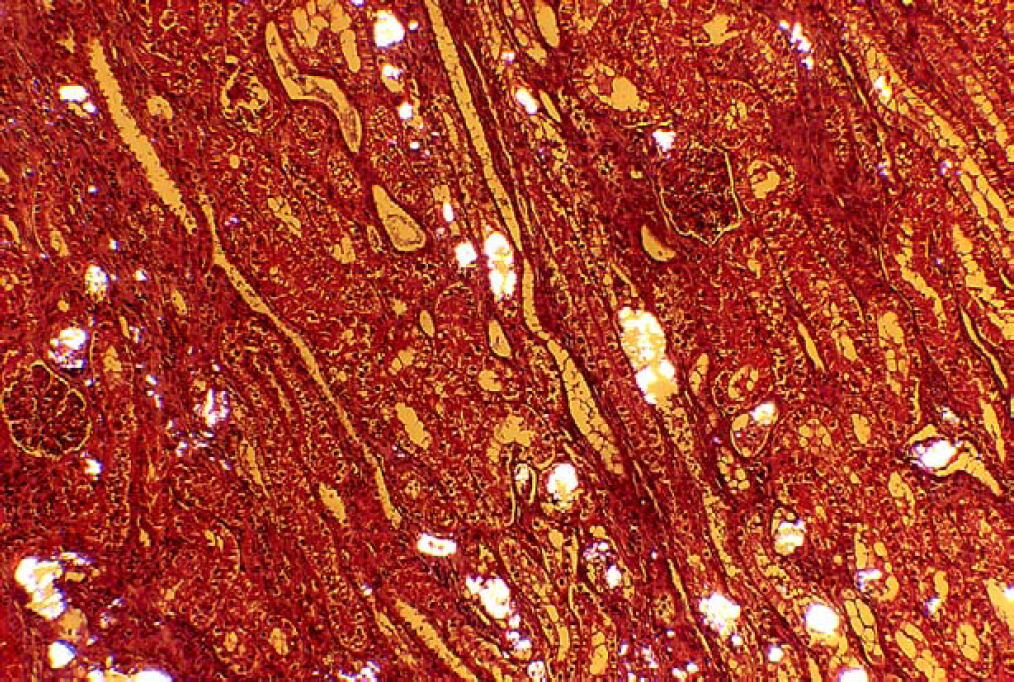
Kidney. Widespread localisation of intraluminal birefringent oxalate crystals under polarised light (haematoxylin and eosin ×100).

Kidney. An oxalate crystal under medium polarised light (haematoxylin and eosin under polarised light ×400).
Primary hyperoxalurias are a group of inherited diseases described in humans, dogs and cats. In humans two different forms of PH are described: type 1 is characterised by associated glycolic aciduria, type 2 by associated l-glyceric aciduria (Petrarulo et al 1998). Type 1 PH, due to a deficiency of the enzyme alanine:glyoxylate aminotransferase (AGT), is clinically characterised by chronic progressive renal failure and neurological signs have been reported. Type 2 PH, due to a deficiency of the hepatic activity of the enzyme d-glycerate dehydrogenase, presents as acute renal failure and has never been described in association with neurological signs. PH in cats, due to a deficiency of the hepatic activity of d-glycerate dehydrogenase, is clinically an intermediate form of the disease, presenting with acute renal failure and neurological signs (McKerrel et al 1989). Pathogenic mechanisms involved in the development of lesions in the nervous system in cats are still unknown.
In the original description (McKerrel et al 1989) the predominant clinical feature was the development of acute renal failure. In our case, while serum and urine analysis was compatible with renal disease, the cat did not have severe renal impairment. In contrast, neuromuscular function was markedly affected. This suggests that involvement of the nervous system may be more severe and present earlier than the renal disease in our case.
Calcium oxalate monohydrate crystalluria may be primary or may occur secondary to ethylene glycol poisoning (Thrall et al 1984), specific plant intoxication (Dickie et al 1978) or low intake of piridoxine (Di Tommaso et al 2002). This was an indoor cat, fed with a balanced commercial cat food and nothing in the history suggested any of these intoxications. For these reasons, the intermittent detection of oxalate in the urine and the neurological signs, primary hyperoxaluria was suspected and specific urine analysis for metabolically related hyperacidurias were performed.
Pathologically, the most important difference between humans and cats with PH is that neurofibrillary accumulation has not been described in humans while it is common in the feline species (McKerrel et al 1989). While neurofibrillary accumulation has been demonstrated in other domestic species including rabbits (Shields and Vandevelde 1978), dogs (Shell et al 1987, Cork et al 1990), pigs (Higgins et al 1983), cattle (Rousseaux et al 1985, El-Hamidi et al 1989, Agerholm and Basse 1994, Pumarola et al 1997), feline primary hyperoxaluria is the first congenital feline disease where neuronal neurofilament accumulation is one of the major neuropathological features. Other descriptions of feline disorders characterised by axonal neurofilament accumulation have an adult onset (Shelton et al 1998). In 1976, Vandevelde et al described a case of a cat that shares clinical and pathological features with PH, but the aetiology could not be determined. As the authors hypothesised a congenital metabolic disease and in the following years no other congenital feline diseases characterised by neurofilaments accumulation have been described, it is possible that this cat had PH.
McKerrel et al (1989) proposed the cat as a model for human hyperoxaluria and human motor neuron diseases (amyotrophic lateral sclerosis) and the cat could provide a useful model to assess enzyme inhibitors which reduce the formation of oxalate.
In many of the congenital neurological diseases in animals the aetiology is unknown (Summers et al 1995). Inborn metabolic abnormalities are often suspected, but are not supported by routine blood analyses and urinalysis. In the recent years an increasing number of case reports (Podell et al 1996, Morgan and McConnell 1999) on acidurias in dogs with neurological diseases have been reported. In these cases in dogs as in the cat of our case, the finding of aciduria allowed an early diagnosis. Therefore, the search of by-products indicative of enzyme dysfunction by urinalysis ought to be performed in the work-up of congenital neurological diseases, as it might help in identifying a potential aetiology and thus understanding of the pathogenesis.
Acknowledgement
The authors thank Dr André Jaggy for reading the manuscript and for his useful suggestions.