
Abstract
16S rRNA gene sequence analysis provided evidence for two different mycobacterial species, Mycobacterium lepraemurium and a potentially novel species, as causative agents of ‘feline leprosy’. Comparison of 16S rRNA gene sequence data obtained for M. lepraemurium and the potentially novel species indicated 12 nucleotide differences over a 446 bp region encompassing the V2 and V3 hypervariable regions. From available 16S rRNA gene sequence data, M. lepraemurium shared greatest nucleotide identity with M. avium subsp paratuberculosis and M. avium. The novel species had a long helix 18 in the V3 region and shared greatest nucleotide identity with M. leprae, M. haemophilum and M. malmoense. The novel species had an additional ‘A’ nucleotide at position 105 of the aligned 16S rRNA gene sequence, the only other mycobacterial database sequence having this same extra nucleotide being M. leprae. This nucleotide variation was exploited to develop specific PCR assays for the two species. These were found to be effective and specific when tested against a panel of mycobacteria including species found in feline leprosy lesions and closely related mycobacteria and also when applied directly to formalin-fixed, paraffin-embedded tissues from feline leprosy cases.
Feline leprosy is a disease in which cats develop granulomas in the skin or subcutis. Granulomas contain acid-fast bacilli (AFB) which usually do not grow using standard mycobacteriological methods (Pedersen, 1988). The disease is geographically widespread (Malik et al., 2002; Pedersen, 1988) and Mycobacterium lepraemurium, a slow growing mycobacterial species that is very difficult to culture (Banerjee et al., 1995; Jenkins et al., 1992; Kato and Gozsy, 1966; Pattyn and Portaels, 1980; Rees and Tee, 1962), has been traditionally considered to be the causative agent (Leiker and Poelma, 1974).
Recently, a re-appraisal was made of 13 cases diagnosed presumptively as feline leprosy in Australia. This resulted in the proposal that feline leprosy comprises two syndromes, one associated with M. lepraemurium and another with a potentially novel, fastidious, slow-growing mycobacterial species (Malik et al., 2002). Recent work from North America has indicated that additional slow growing mycobacterial taxa (provisionally called M. visibilis) may cause disseminated disease in cats but in association with diffuse (rather than nodular) cutaneous lesions (Appleyard and Clark, 2002).
The Australian data suggested that M. lepraemurium gave rise to initially localised, but rapidly progressive disease, typically in young cats and associated with a tuberculoid host response, caseous necrosis and gross ulceration of lesions. The novel species, in contrast, was associated with slowly progressive disease of aged cats associated with a lepromatous response, but neither necrosis nor ulceration. Disease associated with M. lepraemurium was considered to be a primary infection of an immunocompetent host by mycobacterial species that cause systemic disease in rats and therefore are likely to have inherent pathogenicity, at least for some cats. In contrast, disease due to the novel species was thought to represent a disseminated opportunistic infection of cats rendered immunodeficient by factors such as long-standing FIV infection, old age or chronic renal insufficiency. Morphological features including the size of mycobacteria and their propensity to take up haematoxylin stain could be used also to help distinguish between M. lepraemurium and the novel species, but were not definitive.
With one exception, culture from all 13 Australian cases had been unsuccessful, despite the use of a range of appropriate media and the presence of abundant AFB in most specimens. Consequently PCR analysis, using universal primers to amplify 16S rRNA gene sequence encompassing the V2 and V3 hypervariable regions (Kempsell et al., 1992), was performed on 11 of the Australian cases. These regions are of particular value for molecular differentiation of mycobacterial species (Rogall et al., 1990a). Specimens included formalin-fixed, paraffin-embedded, fresh and freeze-dried biopsies, and utilised different methodologies in the two participating laboratories (Malik et al., 2002). Three distinct sequences, M. lepraemurium (Fig. 1) (EMBL accession no.: AJ279017; 446 bp), the potentially novel mycobacterial species (Fig. 1) (EMBL accession nos.: AJ294740-6; 556 bp) and a mycobacterial species that did not match any database sequence, were obtained. The latter sequence was found in one case only, a 2-year-old cat with numerous AFB in its lesions. PCR products could not be obtained for two cases.

Manual alignment of 16S rRNA gene sequences of M. lepraemurium (AJ279017) and mycobacterial species (AJ294746) with closely related mycobacterial species; M. malmoense (EMBL accession no. AF152560), M. avium (EMBL accession no. X52918), M. avium subsp paratuberculosis (EMBL accession no. X52934) and M. leprae (EMBL accession no. X53999) and also with M. bovis (EMBL accession no. X52917). The first nucleotide corresponds to nucleotide 21 of aligned mycobacterial 16S rRNA gene sequences (Rogall et al., 1990b). Primer sequences for the species-specific PCR assays are underlined. Variable regions V1, V2 and V3 are highlighted. Symbols: periods, no change; ∗, nucleotide sequence not determined; –, gaps were introduced to obtain maximum levels of nucleotide identity.
The M. lepraemurium sequence, previously detected in four specimens from cats in New Zealand (Hughes et al., 1997), was identified in two of the Australian specimens. The mycobacterial sequence (AJ294740-6), originally detected in two New Zealand specimens, and thought to represent a potential novel mycobacterial species (Hughes et al., 1997), was detected in six of the Australian specimens. The novel mycobacterial species (AJ294740-6) was identified exclusively in older Australian and New Zealand cats (9 to 14 years), whereas, M. lepraemurium, with one exception (a New Zealand cat of 8 years), was detected in young cats (1 to 3 years-of-age).
This communication reports on analyses of a further putative feline leprosy specimen (formalin-fixed, paraffin-embedded) from a 2-year-old French cat and an additional fresh biopsy from another Australian cat (9 years-of-age) with feline leprosy using the same 16S rRNA gene PCR assays with ‘universal’ primers and sequence analyses. Both specimens contained numerous AFB. Analysis of these additional cases and previous studies (Hughes et al., 1997; Malik et al., 2002) enabled comparisons of 16S rRNA sequences from a diverse range of feline leprosy cases to be made. Observations concerning sequence data obtained for the two major mycobacterial species identified in feline leprosy cases and the development of two PCR assays to enable detection and differentiation of these aetiological agents are reported.
Materials and methods
Isolates
The following mycobacterial strains were obtained from the National Collection of Type Cultures, PHLS Central Public Health Laboratory, London, UK; M. avium (NCTC 8559), M. bovis (NCTC 5693), M. fortuitum (NCTC 10394), M. intracellulare (NCTC 10425), M. kansasii (NCTC 10268), M. marinum (NCTC 10009), M. scrofulaceum (NCTC 10803), M. terrae (NCTC 10856), M. xenopi (NCTC 10042). Mycobacterial type strains were cultured on Lowenstein-Jensen slopes with pyruvate and subcultured in Middlebrook 7H9 broth medium (Difco Laboratories, USA) at 37°C. Field strains of Mycobacterium avium subsp paratuberculosis and Mycobacterium malmoense were isolated from cattle by culture in BACTEC 12B medium (Becton Dickinson, UK) and subcultured in Middlebrook 7H9 broth medium at 37°C. Cloned 16S rRNA PCR products of M. lepraemurium and the novel mycobacterial species (AJ294746), obtained from New Zealand feline leprosy cases 5 and 19 respectively (Table 1), were cloned as described previously (Hughes et al., 1997), and E. coli transformants were subcultured in Luria-Bertani (LB) broth at 37°C.
Age of cat, country of domicile, presence of AFB and mycobacterial species identified in putative feline leprosy specimens
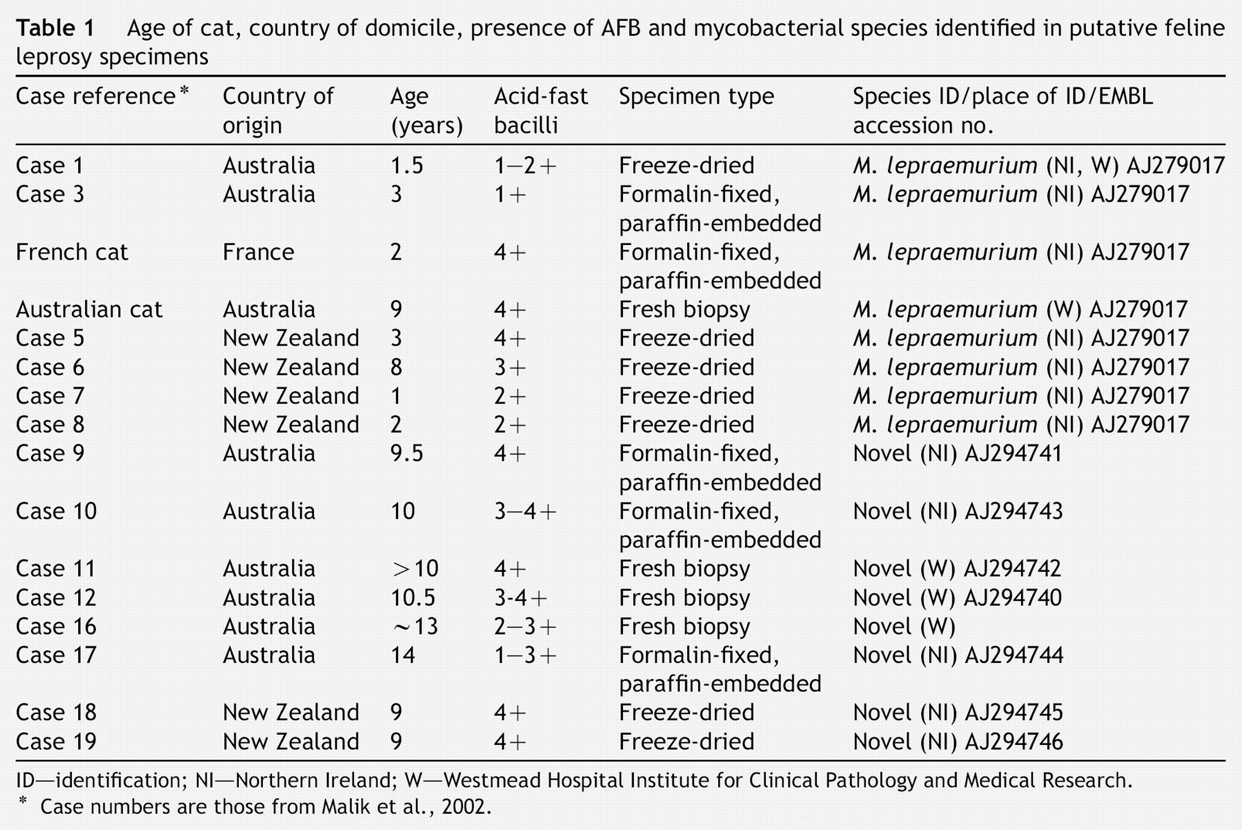
ID—identification; NI—Northern Ireland; W—Westmead Hospital Institute for Clinical Pathology and Medical Research.
Case numbers are those from Malik et al., 2002.
Extraction of DNA for PCR amplification
DNA was extracted from mycobacterial cultures and from LB broth cultures of cloned 16S rRNA amplicons by a simple heat treatment method. For each culture, a 1 ml aliquot was pelleted by centrifugation at 13,000 g for 5 min. The pellet was resuspended in 100 μl sterile deionised water and heat treated at 100°C for 10 min. 16S rRNA gene sequences were extracted from formalin-fixed, paraffin-embedded specimens from Australian cases 3, 10 and 17 and from the French cat (Table 1) by a DNA sequence capture method using biotinylated capture oligonucleotides pA (5′ AAAAAAGAGTTTGATCCTGGCTCAG) and pH (5′ AAAAAAAGGAGGTGATCCAGCCGCA) as described previously (Hughes et al., 2000). DNA was extracted from the fresh biopsy specimen of the further Australian cat (Table 1) in the laboratory at Westmead using a sodium dodecyl sulphate (SDS)-mediated cell lysis, phenol-choroform extraction and ethanol precipitation as described previously (Hughes et al., 2000).
PCR amplification
Universal 16S rRNA PCR amplifications, encompassing the V2 and V3 variable regions, were performed on extracted DNA to identify the AFB observed in the French and further Australian cat specimens as described (Hughes et al., 2000). Sequence analysis of 16S rRNA PCR amplicon from the French cat was performed by the Advanced Biotechnology Centre, London, UK and the amplicon obtained for the Australian cat was sequenced as described (Hughes et al., 2000). Resulting sequences were analysed using Chromas version 1.42 (Griffith University, Brisbane, Queensland, Australia) and DNASIS version 7 software (Pharmacia Biosystems GmbH). Consensus sequences were compared with EMBL, GenBank and DDJB databases using the UK MRC HGMP-RC BLAST programme.
16S rRNA PCR assays were developed for the specific detection of M. lepraemurium and the novel mycobacterial species (AJ294740-6). For M. lepraemurium, the nucleotide sequences of the forward primer (22 mer) and reverse primer (21 mer) were 5′ gaatattgcacaatgggcgcag 3′and 5′ aaacccggaccttcgtcgata 3′ respectively (Fig. 1) and the length of the anticipated PCR product was 102 bp. For the novel mycobacterial species, the nucleotide sequences of the forward primer (21 mer) and reverse primer (20 mer) were 5′ caggga-taagcctgggaaacc 3′and 5′ acccgaaggccgtcatccct 3′ respectively (Fig. 1) and the length of the anticipated PCR product was 284 bp. For both PCR assays annealing temperatures of 60°C, 65°C, 68°C and 70°C with final MgCl2 concentrations of 1.5 mM and 1.75 mM were tested initially to optimise PCR conditions. DNA extract from undiluted heat treated cloned PCR product and plasmid preparations of cloned PCR product prepared previously (Hughes et al., 1997) from cases 5 (M. lepraemurium) and 19 (novel mycobacterial species AJ294746) were used in these initial experiments and the PCR amplifications were performed in duplicate.
After preliminary optimisation experiments, 5 μl DNA extract was added to 45 μl PCR mix containing forward and reverse primers (30 pmol of each), MgCl2 (1.5 mM final concentration), 1.25 units of HotStar Taq DNA polymerase (Qiagen Ltd, UK), 5 μl X10 PCR buffer (Qiagen Ltd) and deoxyribonucleotides (each at a final concentration of 200 nM). Thermocycling conditions were 15 min at 94°C, followed by 40 cycles of 1 min at 94°C, 1 min at 70°C, 1 min at 72°C and a final extension period of 7 min at 72°C. The specific PCR assays were performed on all the mycobacterial type cultures and field strains in duplicate. In addition, both PCR assays were applied to DNA extracted by sequence capture from case 3 and the French cat in which M. lepraemurium had been identified previously and from cases 10 and 17 in which the novel mycobacterial species (AJ294743 and AJ294744) had been identified previously. Cloned PCR amplicons from cases 5 and 19 were used as positive and negative controls for each specific assay and negative controls for the PCR mix were included. PCR amplifications of sequence capture material were subjected to a second round of amplification in accordance with two rounds of amplification being used for the universal 16S rRNA PCR amplifications. The thermocycling conditions for the second round of amplifications were as before but the number of cycles was reduced to 25 and 2 μl instead of 5 μl inoculum was used. Amplicons (5 μl) were visualised by electrophoresis on agarose gels stained with ethidium bromide.
Results and discussion
PCR amplification and sequence analysis of 16S rRNA gene sequences identified M. lepraemurium in the French feline leprosy specimen and interestingly, also in the Australian specimen, despite the latter originating from an older cat. From this study and previous work (Malik et al., 2002) partial 16S rRNA gene sequences, identified as M. lepraemurium, were obtained from a total of eight specimens of diverse geographical (Australia, New Zealand and France) and temporal origin. All eight sequences, determined in two different laboratories, were identical to each other. From the previous study, seven sequences (AJ294740, 1, 2, 3, 5, 6 and an unsubmitted sequence), again determined in two different laboratories, were obtained for the potentially novel mycobacterial species and were identical to each other. A further sequence for the potentially novel mycobacterial species (AJ294744), had a ‘T’ nucleotide at position 423 of aligned mycobacterial 16S rRNA gene sequences (Rogall et al., 1990b), within the V3 variable region, whereas the others had a ‘C’ nucleotide at this position. The nucleotide difference was considered to be neither a sequencing artefact nor due to an error generated by the Taq polymerase since sequencing was performed directly on both strands of amplicons generated on different occasions without recourse to cloning. All eight sequences were characterised by a long helix 18 in the V3 region, indicating that the potentially novel mycobacterial species is likely to be a slow grower (Springer et al., 1996). This accords with the one case cultured successfully which grew very slowly on Lowenstein-Jensen medium (supplemented with iron) and semisolid agar (Malik et al., 2002). Comparison of the 16S rRNA gene sequence data obtained for M. lepraemurium and the potentially novel mycobacterial species indicated 12 nucleotide differences (13 for AJ294744) over 446 bp of the 16S rRNA gene.
From available 16S rRNA gene sequence data, M. lepraemurium shared greatest nucleotide identity (99.3% over 446 bp) with M. avium subsp paratuberculosis (EMBL accession no. X52934) and (99.1% over 446 bp) with M. avium (EMBL accession no. X52918). Mycobacterial sequence (AJ294746) shared highest nucleotide identities with the following mycobacterial species; M. leprae, M. haemophilum and M. malmoense (Table 2). Comparison of novel mycobacterial sequence AJ294746 with recently obtained sequence data, Mbv1, Mbv2 and Mbv3, from three North American cases of ‘feline multisystemic granulomatous mycobacteriosis’ (Appleyard and Clark, 2002) were made (Table 2) and indicated that they were not the same.
Percentage nucleotide identity of potential novel mycobacterial species (AJ294746) to mycobacterial species sharing highest nucleotide identity and to sequences from North American cases of feline multisystemic granulomatous mycobacteriosis

The 16S rRNA gene sequence of the novel mycobacterial species (AJ294740-6) had an additional ‘A’ nucleotide at position 105 of aligned 16S rRNA gene sequences for mycobacterial species (Rogall et al., 1990b). The nucleotide had not been included in the partial 16S rRNA gene sequence presented earlier for the respective New Zealand feline leprosy specimens (Hughes et al., 1997), although it was present in both sequences. The additional nucleotide in these sequences was considered, at that time, to be a sequencing artefact. Interestingly, the only other mycobacterial database sequence associated with the same extra nucleotide is M. leprae (B. Springer, personal communication). This nucleotide was not associated with the North American Mbv1, 2 or 3 sequences. Like M. leprae and Mbv1, Mbv2 and Mbv3, the novel mycobacterial species (AJ29470-6) has an insert in the V1 region (Fig. 1). Human leprosy, caused by M. leprae, is associated with invasion of local nerves and although this is not reported to occur commonly in feline leprosy, occasional cases from North America have documented infiltration of peripheral nerves with AFB (Matthews and Liggitt, 1983; Paulsen et al., 2000).
In the re-appraisal of Australian feline leprosy specimens, PCR amplification and analysis of 16S rRNA gene sequences enabled reclassification of the presumptive feline leprosy cases into two groups. Although the two clinical syndromes could generally be distinguished by taking clinical and microscopic features into account, only molecular analyses distinguished the two syndromes unequivocally. Morphologic and staining characteristics are not specific and disadvantaged by subjectivity. Moreover, epidemiologic factors and knowledge of the host immune response may be required to distinguish M. lepraemurium infections from those caused by the novel species. For example, the propensity of M. lepraemurium infections to occur in young cats likely reflects the greater tendency for young cats to hunt rodents. Thus, it is hardly surprising that the occasional older cat will develop a M. lepraemurium infection as a result of an altercation with an infected rat. Likewise, the lepromatous response seen typically in infections caused by the novel species probably reflects immunodeficiency per se rather than a feature of the causal organism (Bartralot et al., 2000). It is therefore conceivable that in such a host, M. lepraemurium may be associated with lepromatous pathology rather than the tuberculoid response typically seen in immunocompetent young cats.
Thus although routine diagnosis of feline leprosy is considered to be straightforward (Malik et al., 2002), definitive identification can only be reliably provided by molecular analyses. Such a precise diagnosis is useful because different approaches to therapy, especially in relation to the requirement for surgery, have been recommended for different cutaneous mycobacterial syndromes (Malik et al., 2002). A further issue is the need to rapidly distinguish the agents of feline leprosy from mycobacterial species that pose a zoonotic threat to humans. Slow-growing mycobacterial species such as M. microti (Gunn-Moore et al., 1996; Kremer et al., 1998), M. avium (Hughes et al., 1997) and M. genavense (Hughes et al., 1999) have been isolated from presumptive feline leprosy cases, although typically these cases have systemic involvement in addition to skin lesions. Likewise, diagnosis based solely on histological analyses has been thought to account for cats infected with M. bovis being misdiagnosed as having feline leprosy (de Lisle et al., 1990). Although culture is possible in these cases, molecular analyses provide a more rapid diagnosis, which is particularly important for cat owners when zoonotic pathogens such as M. bovis and M. microti are potentially involved. With the advent of commercial DNA probe kits and microarrays relevant for veterinary diagnostics, it should be feasible to analyse PCR products with more rapidity than current ‘in-house’ reverse cross blot hybridisation assays and sequence analyses of 16S rRNA gene amplicons allow. However, in the interim, a more practical approach for definitive diagnoses of feline leprosy syndromes may be to utilise specific PCR tests for the main bacterial species identified.
This communication reports the development of two PCR assays based on the 16S rRNA gene for the major aetiological agents of feline leprosy identified by Malik et al. (2002). The 16S rRNA gene is highly conserved but has regions of high variability, which are species-specific for mycobacteria (Rogall et al., 1990a). Sequence comparisons indicated that the 16S rRNA gene sequences of M. avium and M. avium subsp paratuberculosis are identical to M. lepraemurium in the V2 region. However, there are a few nucleotide differences between the 16S rRNA gene sequences of M. avium and M. avium subsp paratuberculosis and that of M. lepraemurium over the 446 bp region of comparison (Fig. 1). This indicated the potential for development of a M. lepraemurium specific PCR assay. The primers were designed so that each nucleotide difference formed the 3′ end of each primer.
Similarly, primers were designed for a 16S rRNA gene PCR assay specific for the novel mycobacterial species (AJ294740-6) found in older cats. Again, the specificity of the assay relied on single nucleotide mismatches at the 3′ end of each primer to differentiate this species from all other mycobacterial species for which 16S rRNA gene sequence information was available from public databases. The primers were designed to amplify regions within the 16S rRNA gene which were identical for all eight sequences obtained from the variety of feline leprosy specimens in which the potential novel mycobacterial species was identified.
Preliminary optimisation experiments revealed that both PCR assays were effective and specific at an annealing temperature of 70°C but not at the other annealing temperatures tested, although only very faint bands were associated with non-specific amplification at 68°C and these were not present in all of the amplifications. The intensities of PCR amplicons for the potentially novel pathogen were relatively stronger than those for M. lepraemurium despite the latter being smaller. However, the PCR amplicons for M. lepraemurium were clearly visible despite the high stringency of the PCR assay and there is potential to modify primers by extending one or both of the primers at the 5′ end, whilst retaining the 3′ end mismatches, and PCR conditions to optimise the amplification. The specificity of each assay was tested against a range of mycobacterial species. These included mycobacterial species sharing highest nucleotide identity with either M. lepraemurium or the novel mycobacterial species (AJ294746), mycobacterial species which may be associated with presumptively diagnosed feline leprosy cases and other mycobacterial species. M. microti was not included since the 16S rRNA sequence encompassed by the primers for this species is identical to that of M. bovis, both species being members of the tubercle complex. Each assay was specific for the mycobacterial species it was developed for and PCR amplicons were not obtained for any of the 12 other mycobacterial species tested (Fig. 2).
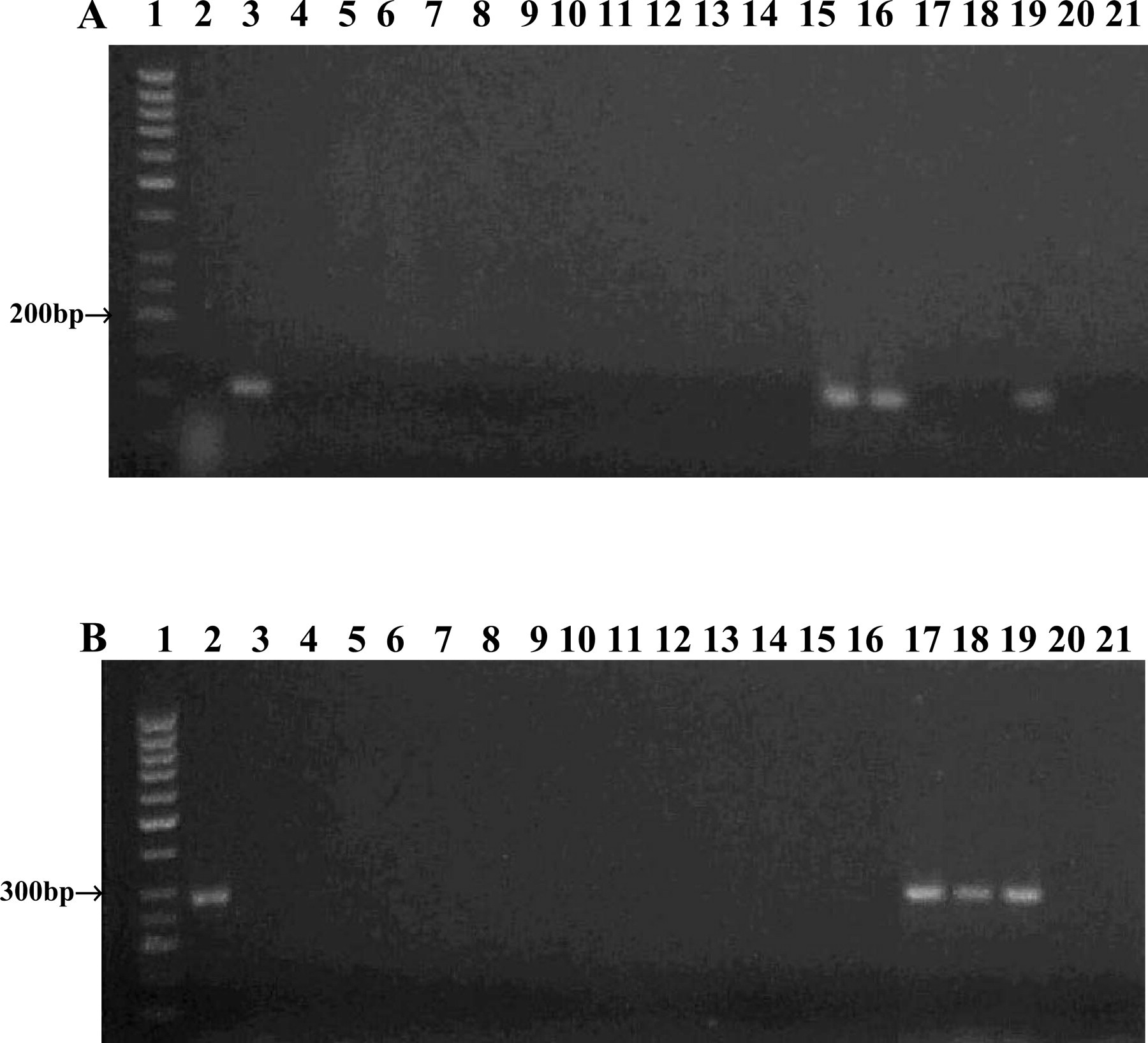
PCR amplification of DNA extracted from feline leprosy specimens and a panel of mycobacterial species using A) the M. lepraemurium assay and B) the assay for the novel mycobacterial species. PCR products were visualised on a 2% (w/v) agarose gel by ethidium bromide staining. Lane 1; 50 bp DNA ladder, lane 2; cloned 16S rRNA PCR product of the novel mycobacterial species (AJ294746), lane 3; cloned 16S rRNA PCR product of M. lepraemurium, lane 4; M. avium (NCTC 8559), lane 5; M. bovis (NCTC 5693), lane 6; M. fortuitum (NCTC 10394), lane 7; M. intracellulare (NCTC 10425), lane 8; M. kansasii (NCTC 10268), lane 9; M. marinum (NCTC 10009), lane 10, M. scrofulaceum (NCTC 10803), lane 11; M. terrae (NCTC 10856), lane 12; M. xenopi (NCTC 10042), lane 13; M. avium subsp paratuberculosis (field strain), lane 14; M. malmoense (field strain), lane 15; feline leprosy case 3R, lane 16; French catR, lane 17 feline leprosy case 17R, lane 18 feline leprosy case 10R, lane 19A; cloned 16S rRNA PCR product of M. lepraemurium R, lane 19B; cloned 16S rRNA PCR product of the novel mycobacterial species (AJ294746)R, lane 20; PCR mix inoculated with water, lane 21 PCR mix inoculated with waterR. R=second round of amplification.
Application of both assays to clinical specimens was effective and specific also. The assay for M. lepraemurium yielded amplicons of expected size (102 bp) for feline leprosy case 3 and the French cat in which M. lepraemurium had been identified previously, but did not amplify DNA extracted by sequence capture from feline leprosy cases 10 and 17 in which the novel mycobacterial pathogen had been identified. Conversely, the assay for the novel pathogen yielded amplicons of expected size (284 bp) for feline leprosy cases 10 and 17 but not for feline leprosy case 3 or the French cat (Fig. 2). Amplicons were obtained from the formalin-fixed, paraffin-embedded specimens after the first round of amplification as well as after the second round. Although the intensity of M. lepraemurium amplicons was stronger after the second round of amplification, two rounds of amplification may not be necessary. For both assays, the negative controls for both rounds of amplification (including a reamplified negative control) did not yield amplicons. For future diagnostic applications of these assays to clinical specimens it is recommended that a sequence capture methodology be used for DNA extraction to remove potential inhibitors including tissue DNA. DNA, extracted by sequence capture, could be subjected to three separate PCR assays, the two specific PCR assays for M. lepraemurium and the novel mycobacterial species, and a universal 16S rRNA PCR assay encompassing the V2 and V3 variable regions. The latter would ensure against false negatives in the specific assays and provide amplicon for sequence analysis if the specimen does not contain M. lepraemurium or the novel mycobacterial species.
Subsequent to the development of the described PCR assays, three cases of feline multisystemic granulomatous mycobacteriosis from different regions in the USA and Canada were reported (Appleyard and Clark, 2002). Analysis of the sequence data provided for the three cases revealed that the primer set developed for the novel mycobacterial species (represented by AJ294740-6) in Australian and New Zealand cats, should, in theory, detect the sequences for the North American ‘M. visibilis’ sequences Mbv1 and Mbv2 despite the novel mycobacteria species (AJ294746) differing from these sequences by 20 and 19 nucleotides over 543 bp, respectively. The primers for the novel mycobacterial species (AJ294740-6) should not detect the North American sequence Mbv3 using the PCR conditions described. The 284 bp amplicons of the novel mycobacterial species (AJ294740-6) could be easily differentiated from amplicons of Mbv1 and Mbv2 by a simple restriction enzyme digest. BsmaI should cut the PCR products of Mbv1 and Mbv2 once to generate two fragments of 45 bp and 239 bp, but not the amplicons of the novel species (AJ294740-6).
Increased application of molecular analyses to feline mycobacteriosis, resulting in the identification of new fastidious mycobacteria, has highlighted the need for a molecular approach to definitively diagnose feline mycobacterial disease entities. In this study two PCR assays, based on 16S rRNA gene sequences, have been developed. These PCR assays should allow rapid, sensitive and specific diagnosis of the pathogens from specimens of presumptive feline leprosy cases, without the requirement for culture. Furthermore, the provision of these molecular tools will facilitate future studies of the aetiopathogenesis of these infections, for example by permitting a search for the environmental niche of causal organisms and the participation of postulated mechanical vectors.
Footnotes
Acknowledgement
We wish to thank G. de Lisle, AgResearch, Wallaceville Animal Research Centre, Upper Hutt, New Zealand and N. Haddad, Ecole Nationale Vétérinaire d'Alfort, Maisons Alfort Cedex, France for the New Zealand and French feline leprosy specimens respectively and relevant information. Vanessa Barrs provided the most recent Australian specimen. We would also like to thank B. Springer and E.C. Böttger, Institute für Medizinische Mikrobiologie, Medizinische Hochschule Hannover, Germany for their valuable comments regarding the 16S rRNA gene sequence of the mycobacterial species (AJ294746). This paper is dedicated to the memory of Daria Love whose enthusiasm and commitment inspired this work. This work was supported by a grant from the Australian Companion Animal Health Foundation.