
Abstract
Traditionally, G protein-coupled receptors (GPCRs) were thought to function as monomeric units activating linear signaling pathways to reach a single functional response. However, it is now recognized that GPCRs can exist as higher order structures, such as homomers or heteromers. The potential for unique pharmacology attributed to these GPCR complexes has opened up the possibility of a new class of targets that can be exploited for drug discovery. In this innovation brief, a novel technology developed to identify and profile GPCR heteromers and their ligands will be reviewed.
Introduction
G protein-coupled receptors (GPCRs) are the largest family of cell surface receptors. By responding to a vast array of stimuli, such as sensory messages (photons and odors) and messenger molecules (such as biogenic amines, lipids, and peptides), GPCRs allow cells to communicate with each other and their surroundings. Widely expressed in the human body, GPCRs are implicated in a wide range of disease pathways, including cancer, cardiomyopathy, and schizophrenia. Around 50% of the drugs on the market regulate GPCR function, 30% of which act directly on GPCRs. 1 Despite this, however, of the 800 or so receptors that make up this superfamily, drugs modulating GPCR function only act on approximately 30 receptors. 2 This knowledge, taken together with their diverse physiological and pathological roles, highlights the potential for drug discovery in this field.
The belief that GPCRs act as isolated monomeric entities has been challenged for nearly 30 years (see ref 3 for review), and it is now widely accepted that not only can GPCRs interact with other GPCRs to form higher order structures but also such complexes can exhibit distinct pharmacology from their respective protomers. This pharmacology has been termed the “biochemical fingerprint.” 4 The numerous potential heteromer combinations and the opportunity for tissue-specific expression present the exciting possibility of designing drugs with greater selectivity and reduced side effects. It is therefore, no surprise that GPCR heteromerization has attracted interest from both academic and pharmaceutical industry researchers. To achieve this specificity and selectivity via heteromer signaling, it is crucial to first develop assays that can distinguish signaling from monomer/ homomers and heteromers. It is then important to identify ligands that selectively act on the heteromer of interest or at least result in distinct heteromer-biased pharmacology (see ref 5 for review).
G Protein-Coupled Receptor Heteromer Identification Technology
The G protein-coupled receptor heteromer identification technology (GPCR-HIT) is a novel approach to identify and profile heteromers and the ligands that act on them.5–7 This technology is based on a configuration made up of three elements, only two of which are tagged with complementary components of a proximity-based reporter system; labeled GPCRA, untagged GPCRB, and a labeled GPCR-interacting Proteinc (e.g., [β-arrestin or G protein), which is recruited in a ligand-dependent manner (Fig. 1). By applying a ligand selective for untagged GPCRB, the GPCRA-GPCRB heteromer can be monitored by the signal generated when Proteinc is recruited, thereby bringing the label on GPCRA into close proximity with the label on Proteinc. As a signal is only detected as a result of heteromerization and not homomerization, this assay avoids the “noise” that can be an issue when trying to differentiate signals originating from heteromers or homomers.
The GPCR-HIT uses cotransfection of a labeled GPCRA, unlabeled GPCRB, and a labeled Proteinc (such as (β-arrestin or G protein) that interacts with GPCRB or the GPCRA-GPCRB heteromer on binding of a ligand selective for GPCRB or the heteromer. The labels on GPCRA and Proteinc are complementary components of a proximity-based reporter system (RCI, reporter component l; RC2, reporter component 2). Reprinted from Current Opinion in Pharmacology, vol 10, Ayoub, M. A.; Pfleger, K. D., Recent advances in bioluminescence resonance energy transfer technologies to study GPCR heteromerization, pages 44-52, Copyright 2010, with permission from Elsevier.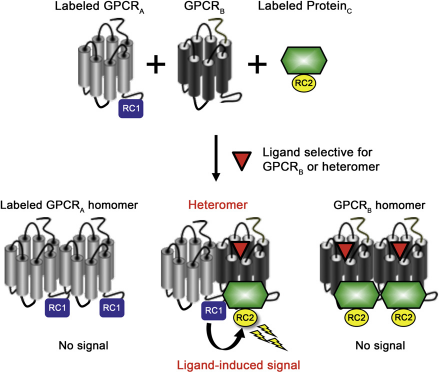
Another benefit of this technology is its adaptability to various assay platforms, such as bioluminescence resonance energy transfer (BRET), fluorescence resonance energy transfer (FRET), bimolecular fluorescence complementation (also known as protein-fragment complementation assay), bimolecular luminescence complementation, enzyme fragment complementation, and the Tango protease-based system. 5 Each assay has its own merits, and assay choice would therefore be dependent on individual experimental requirements, allowing flexibility.
BRET is a biophysical technique, which is increasingly playing an important role in the investigation of protein-protein interactions.
8
Here, a BRET signal can be measured in real time and live cells when two proteins, differentially tagged with either a donor (a Renilla luciferase [Rluc] variant) or an acceptor (a green fluorescent protein variant) are in close proximity (Fig. 2). The resonance energy generated by the oxidation of the coelenterazine substrate by Rluc is transferred to the acceptor only if it is within 10 nm of the donor;
9
the resulting acceptor light emission peaking at a characteristic wavelength can then be measured. When applied to the GPCR-HIT concept, acceptor emission is indicative of the existence of a complex containing both donor- and acceptor-fused proteins.5–7
This schematic drawing illustrates the use of BRET to monitor protein-protein interactions. The resonance energy generated by the oxidation of the coelenterazine substrate by Renilla luciferase variant Rluc8 (the donor) is only transferred to green fluorescent protein variant Venus (the acceptor) if the proteins fused to these components are in close proximity of each other.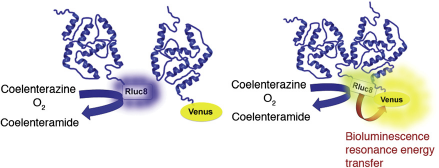
Profiling of Heteromers
The application of GPCR-HIT using BRET has recently been published, profiling the known chemokine receptor heteromer combinations of CCR2-CCR5 and CCR2-CXCR4.
7
By using the multipurpose scaffolding and signaling protein, β-arrestin 2, the specificity of this technology was demonstrated in real time and in live cells. By co-expressing the untagged CCR2 with (β-arrestin 2/Venus and the Rluc8-tagged CCR5 or CXCR4, it was shown that a BRET signal indicative of (β-arrestin recruitment was observed on specific activation of CCR2 by CCL2 (Fig. 3). Furthermore, through the use of real-time kinetic profiles and dose-response curves, potential differences in (β-arrestin recruitment profiles between the heteromer and constituent protomers can be identified.
Kinetic BRET data for GPCR-HIT profiling chemokine receptor heteromerization for CCR2-CCR5 and CCR2-CXCR4. HEK293FT cells transiently co-expressed CCR5/Rluc8 and (β-arrestin 2/Venus in the absence (A) or presence (B) of CCR2. Alternatively, HEK293FT cells transiently co-expressed CXCR4/Rluc8 and (β-arrestin 2/Venus in the absence (C) or presence (D) of CCR2. The cells were monitored at 37 °C using extended BRET to generate kinetic profiles. Selective agonists (alone or in combination; 100 nM) or vehicle were added after 10 min, and measurements recommenced for a further 50 min. Data shown are representative of three independent experiments. Reprinted from ref 7, copyright 2011, with permission from Mary Ann Liebert, Inc.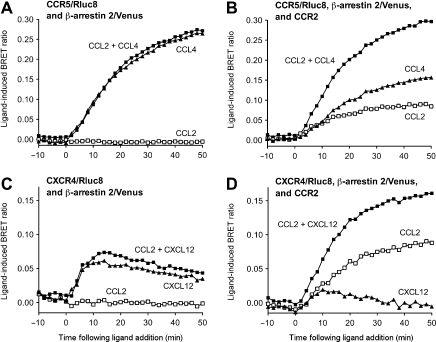
(β-arrestins play an important role in regulating cell signaling and were historically thought of as proteins primarily important for GPCR desensitization and internalization. However, in recent years, it has become clear that these proteins play a critical role in the signaling complexes that ultimately lead to the activation of the members of the mitogen-activated protein kinase family. 10 Evidence in the literature suggests that activation of (β-arrestin and G protein signaling pathways can result in different biological responses. For example, when patients suffering from dislipide-mia are treated with niacin, along with its beneficial lipid lowering effects, patients also experience an unwanted flushing response. 11 Investigation of the mechanism behind these varied responses suggests that agonists acting on the nicotinic acid receptors induce a flushing response only when the (β-arrestin pathway is activated; however, the desired lipid lowering effects have been proposed to be mediated via a G protein-dependent pathway. 12 Therefore, receptor interactions with (β-arrestin resulting in activation of G protein-independent signaling pathways may be exploited or avoided to achieve a desired pharmacological profile.
As most GPCRs interact with (β-arrestins, the aforementioned assay configuration can be applied to many different GPCRs without requiring prior knowledge of the G proteins with which the heteromer interacts, 13 and with the added benefit of using a signaling protein now known to play an important role in GPCR signaling diversity. Indeed, we have highlighted that this assay can also be used in cases where one of the two protomers recruits βarrestin exceptionally poorly when expressed alone but gains the ability to strongly recruit (β-arrestin when in a heteromer complex. Norepinephrine-induced (β-arrestin 2 recruitment to a complex containing the αla-adrenergic receptor (αlaAR) is observed only when this receptor is co-expressed with the chemokine CXCR2 receptor (Mustafa, See, Seeber, Ayoub, and Pfleger, unpublished observations). The α1a-AR monomer/homomer recruits (β-arrestin very weakly, if at all. 14 As discussed earlier, Proteinc can be any protein that interacts with GPCRs in a li-gand-dependent manner. Therefore, in cases where the heteromer complex does not interact with (β-arrestin, tagged G proteins can be used instead. However, as heteromers may exhibit a different G protein preference to their composite receptors, a variety of G proteins may need to be tested.
The advantage of using either (β-arrestin or G protein as Proteinc is the homologous nature of the receptor interaction,
15
with recruitment generally accepted as being either to the activated receptor or an associated protomer within the same macromolecular complex. Indeed, a panel of Rluc8-tagged GPCRs were expressed with (β-arrestin 2/Venus and αlaAR and activated with their cognate agonists. The lack of BRET signal with any of the other GPCRs tested illustrated not only the specificity of the α1aAR-CXCR2 heteromer pharmacology but also the lack of heterologous recruitment of β-arrestin (Mustafa, See, Seeber, Ayoub, and Pfleger, unpublished observations). Another good example that demonstrates the specificity of the GPCR-HIT assay is that of the angiotensin II type 1 receptor (AT1R) and bradykinin type 2 receptor (B2R) combination
7
(Fig. 4). The lack of AngII-induced BRET signal observed when B2R/Rluc8, β-arrestin 2/Venus, and AT1R were co-expressed, despite AT1R normally recruiting (β-arrestin,
16
shows that translocating (β-arrestin to the plasma membrane on activation of any untagged GPCR does not result in a signal simply because of consequent increased proximity with a tagged GPCR at the plasma membrane (Fig. 4). The explanations for the lack of signal with this particular combination include AT1R and B2R not being a heteromer in HEK293FT cells under the experimental conditions used (consistent with ref 17), the AT1R-B2R heteromer having a pharmacological profile with respect to (β-arrestin recruitment that differs from either the constituent monomer/homomer or the B2R/Rluc8-AT1R-β-arrestin 2/Venus complex not being detected as a result of the high sensitivity of BRET to relative donor-acceptor distance and orientation.
7
Lack of GPCR-HIT signal from cells co-expressing B2R and AT|R. Extended BRET (eBRET) at 37 °C was used to generate kinetic profiles with HEK293FT cells transiently expressing B2R/Rluc8 and β-arrestin 2/Venus with or without AT|R (A). Agonist (bradykinin or Ang II; | μM) or vehicle were added after 10 min, and measurements recommenced for a further 50 min. No ligand-induced increase in BRET signal was observed on addition of Ang II(| μM) to B2R/Rluc8 and β-arrestin 2/Venus with or without AT|R (A). Data representative of three independent experiments. To demonstrate plasma membrane expression and functionality of AT|R in this context, the total inositol phosphate production resulting from activation of AT|R with Ang II (| μM) when the cells were co-expressing B2R/RJuc8, β-arrestin 2/Venus, and AT|R was assessed (B). Data shown as fold over basal (total inositol phosphate production from vehicle-treated samples) and are mean ± standard error of the mean of three independent experiments. Z'-factor data were generated using entire 96-well plates of HEK293FT cells transiently co-expressing B2R/RJuc8, β-arrestin 2/Venus, and AT|R (C, D). The plates were monitored at 37 °C using eBRET assaying 48 Ang II (I μM)-treated wells compared with 48 vehicle-treated wells. Fluorescence/luminescence values are presented against time (C) or well number at ∼45 min (D). Solid lines show the means of the positive control (Ang II) and negative control (vehicle). Broken lines display three standard deviations from the mean of each data set. Data representative of three independent experiments. Reprinted from ref 7, copyright 2011, with permission from Mary Ann Liebert, Inc.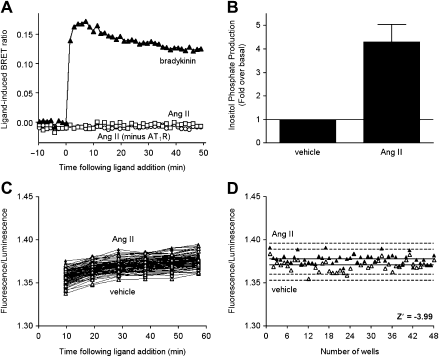
Automation and Validation of Screening Potential
GPCR-HIT requires the transient or stable expression of recombinant proteins, which are labeled with complementary components of a reporter system (Fig. 1). Clearly the use of cell lines stably expressing these proteins is more amenable to automation, although high-throughput transfection can now be achieved using commercially available robotic cell transfection systems, such as the Freedom EVO cell transfection workstation (Tecan, Männedorf, Switzerland). The GPCR-HIT assay is amenable to 96- or 384-well plate formats, the choice of which depends on the underlying technology platform used 5 and whether profiling or screening is being undertaken. Liquid handling to dispense cells, substrate, and/or ligands can be carried out using one of the several liquid handling robotic systems on the market, such as the JANUS automated workstation (PerkinElmer, Glen Waverly, Australia). Assay measurement also depends on the underlying technology platform but generally involves measurement of luminescence and/or fluorescence from adherent or suspended cells using a suitable microplate reader, examples for BRET being the POLARstar Omega, or the LUMIstar Omega (both BMG Labtech, Mornington, Australia), the VICTOR Light (PerkinElmer), or the Mithras LB 940 (Berthold Technologies, Bad Wildbad, Germany). 18 These instruments are able to measure in real time at 37 °C, which may well be desirable. Depending on the level of automation required, substrate and/or ligand injection can be used with such instruments. Furthermore, such microplate readers can be fully integrated with liquid handling workstations so that plates are automatically prepared and measured without manual intervention. Indeed, cell incubators can also be integrated with liquid handing to automate the entire process. When using BRET for GPCR-HIT, there is a choice of substrates available, each resulting in different luminescence decay half-lives. 18 The use of EnduRen (Promega, Sydney, Australia) a protected form of luciferase substrate, can be useful for automation as the prolonged stability of luminescence signal provides additional protocol flexibility. 16 Indeed, real-time extended BRET kinetic profiles have been measured over several hours in N-(2-hydroxyethyl)pipera-zine-N‘-ethanesulfonic acid-buffered media, without requiring CO2 supply to the instrumentation. 16
From a drug discovery perspective, screening is an essential step for identifying potential lead compounds, both at the primary and secondary levels. The screening potential of this technology has been demonstrated by obtaining Z’ values of 0.68 for CCL2-induced signals from cells co-expressing CCR2 with Rluc8-tagged CCR5 or CXCR4
7
(Fig. 5). Z’ values of more than 0.5 indicate high assay performance.
19
There were 720 biologically active compounds screened in a separate study to identify heteromer-biased ligands at the αla AR-CXCR2 heteromer. In addition to a number of known α-adrenergic receptor agonists, a compound previously characterized as a nonselective α/β-AR antagonist was identified as being of interest, and secondary profiling identified novel heteromer-selective pharmacology (Mustafa, See, Seeber, Ayoub, and Pfleger, unpublished observations).
BRET Z'-factor data illustrating the potential for GPCR-HIT screening of chemokine receptor heteromerization for CCR2-CCR5 and CCR2-CXCR4. Entire 96-well plates of HEK293FT cells transiently co-expressing CCR2 and β-arrestin 2/Venus with CCR5/Rluc8 (A, B) or CXCR4/Rluc8 (C, D) were monitored at 37 °C using eBRET assaying 48 CCL2 (100 nM)-treated wells and 48 vehicle-treated wells. Fluorescence/luminescence values are presented against time (A, C) or well number at ∼ 45 min (B, D). Solid lines in B and D show the means of the positive control (CCL2) and negative control (vehicle). Broken lines display three standard deviations from the mean of each data set. Data shown are representative of three independent experiments. Reprinted from ref 7, copyright 2011, with permission from Mary Ann Liebert, Inc.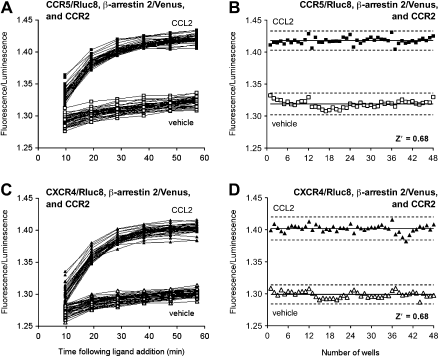
Advantages of GPCR-HIT Compared with other Approaches
One of the main advantages of GPCR-HIT over alternative approaches is that no alteration of GPCR function is required. 5 Although, GPCRA is tagged with a reporter component, this does not alter receptor function, hence allowing the interaction of two receptors to be studied in a system that is closer to the physiological situation. In addition, this technology can be carried out as an end-point assay using a number of proximity-based technologies, or as a kinetic assay, using BRET for example. 5 A live cell-based assay avoids issues of protein precipitation, which is a major downfall of coimmunoprecipitation experiments. 20 By assessing interactions in live cells in a ligand-dependent manner, particularly when using membrane-impermeable ligands, only receptors that are correctly folded and expressed at the cell surface are detected, thus avoiding receptors that are overexpressed/ aggregated in the endoplasmic reticulum. The high sensitivity of the system21,22 enables signals to be monitored using protein expression levels similar to those found endoge-nously; thus, it can be argued that this system provides an intermediate between classical recombinant overexpression studies and in vivo studies.
Traditional FRET and BRET studies to investigate receptor heteromerization involve the measurement of the direct energy transfer between two labeled receptors. As many GPCRs are now thought to exist as constitutive heteromers, 6 thereby being in very close proximity in the absence of ligand, typical BRET or FRET experiments would measure a basal energy transfer that is close to maximal. 20 Therefore, any further ligand-dependent change in energy transfer may not be detected. By using a ligand-dependent GPCR-interacting protein, such as β-arrestin, even constitutive interactions between two receptors can be measured in a ligand-dependent manner. This is important as ligand dependency allows discrimination between a signal because of genuine physical proximity of receptors as opposed to their random collision because of overexpression in recombinant systems. Furthermore, this assay has advantages over BRET saturation assays, 23 which are laborious and not ligand dependent, rendering them unsuitable for even secondary screening. 6
Concluding Remarks
It is now widely accepted that heteromerization can provide a mechanism for modulating signal specificity and diversity, adding another level of “fine-tuning” capability to receptor signaling systems. The GPCR-HIT technology has the potential to play an important role in the identification and profiling of novel heteromers, and by using β-arrestin in particular, has the capacity to identify and investigate novel G protein-independent signaling pathways. It is clearly critical to understand the consequences of receptor heteromerization in physiologically relevant native tissues, and we believe the development of such assays to study GPCR heteromerization in live cells with as little modification of receptor function as possible is an important step toward understanding the complex signaling mechanisms underlying many biological processes in the human body.
Footnotes
Competing Interests Statements: The authors disclose that in addition to being Head of the Laboratory for Molecular Endocrinology—GPCRs, Western Australian Institute for Medical Research and Centre for Medical Research, University of Western Australia, KDGP is Chief Scientific Officer of Dimerix Bioscience Pty Ltd, a spin-out company of the University of Western Australia that has been assigned the rights to the “GPCR-HIT” technology. KDGP has a minor shareholding in Dimerix.
Acknowledgments
Work on GPCR heteromerization carried out in the authors’ laboratory is funded by the National Health and Medical Research Council (NHMRC) of Australia Project Grant #566736, as well as by Dimerix Bioscience Pty Ltd, which has been assigned the rights to the GPCR-HIT technology. KDGP is an Australian Research Council Future Fellow (FT100100271).