
Abstract
Rapid advancement in fluorescence and luminescence imaging technology is encouraging cell biologists to develop high-throughput assays for intracellular detection of bioactive small molecules, protein conformational changes, protein localization and dynamics, and measurement of protein—protein interactions in real time at the level of single living cells with high spatial and temporal resolution. In this report, we summarize the development of genetically encoded fluorescent and bioluminescent probes to study various molecular events in live cells and animals for potential drug candidates against different diseases.
Keywords
Fluorescence resonance energy transfer assays
The extraction of green fluorescent protein (GFP) from the jellyfish, Aequorea victoria, and later the understanding of the chromophore formation, which is responsible for the GFP fluorescence, and GFP cloning have brought a revolution in the field of cell biology. 1 5 The discovery of GFP-like proteins in nonbioluminescent Anthozoa species was another breakthrough in the field of fluorescent proteins (FPs). 6 7 Over the last decade, researchers have contributed a lot toward improving the properties of the GFP through genetic engineering; FPs are developed with diversified spectra, increased brightness and photostability, better folding efficiency, and decreased oligomerization. 8 10 Recently, Roger Tsien laboratory has developed an FP with the longest emission wavelength, 708 nm, called infrared FP, which can be used for deep tissue imaging. 11 The FPs including the photoactivatable 12 are now available for the entire visible spectrum from ∼450 to 710 nm. 13 In most cases, the GFP or GFP-like FP in fusion with a protein of interest does not interfere with the biochemical functions of the protein. 13 14 The choice of FP colors has made fluorescence-based methods including förster (or fluorescence) resonance energy transfer (FRET) microscopy more fascinating to study protein conformational changes and protein—protein interactions in live cells. 15
FRET is a nonradiative transfer of energy from an excited donor fluorophore to an acceptor fluorophore. For FRET to occur, the excitation spectrum of the acceptor must overlap with the emission spectrum of the donor, and the two fluorophores relatively to be close in proximity (< 10 nm) in orientations that permit dipole—dipole coupling. 16 There are two types of FRET, intermolecular and intramolecular. 17 For intermolecular FRET, donor and acceptor fluorophores are fused to two different interacting proteins of interest; in the case of intramolecular FRET, the two fluorophores are attached on two different locations of a single host protein or a protein that may be a concatenation of interacting domains (Fig. 1). The intermolecular FRET allows the monitoring of protein—protein interactions; the FRET signal increases on association of the two proteins, but on dissociation of the proteins, the FRET signal decreases or disappears. The intramolecular FRET measures protein conformational changes, which result in changes in the distances and/or the orientation of the two fluorophores. Although probe molecules based on intramolecular FRET sometimes are difficult to design, it offers several advantages: the donor and acceptor fluorophores are always expressed at a 1:1 stoichiometry, and the ratio of donor to acceptor fluorophore is constant at every pixel; therefore, a simple donor/acceptor (or acceptor/donor) ratio suffices to represent FRET. Moreover, any change in the protein localization and fluorescence bleaching will affect both fluorophores at the same time.
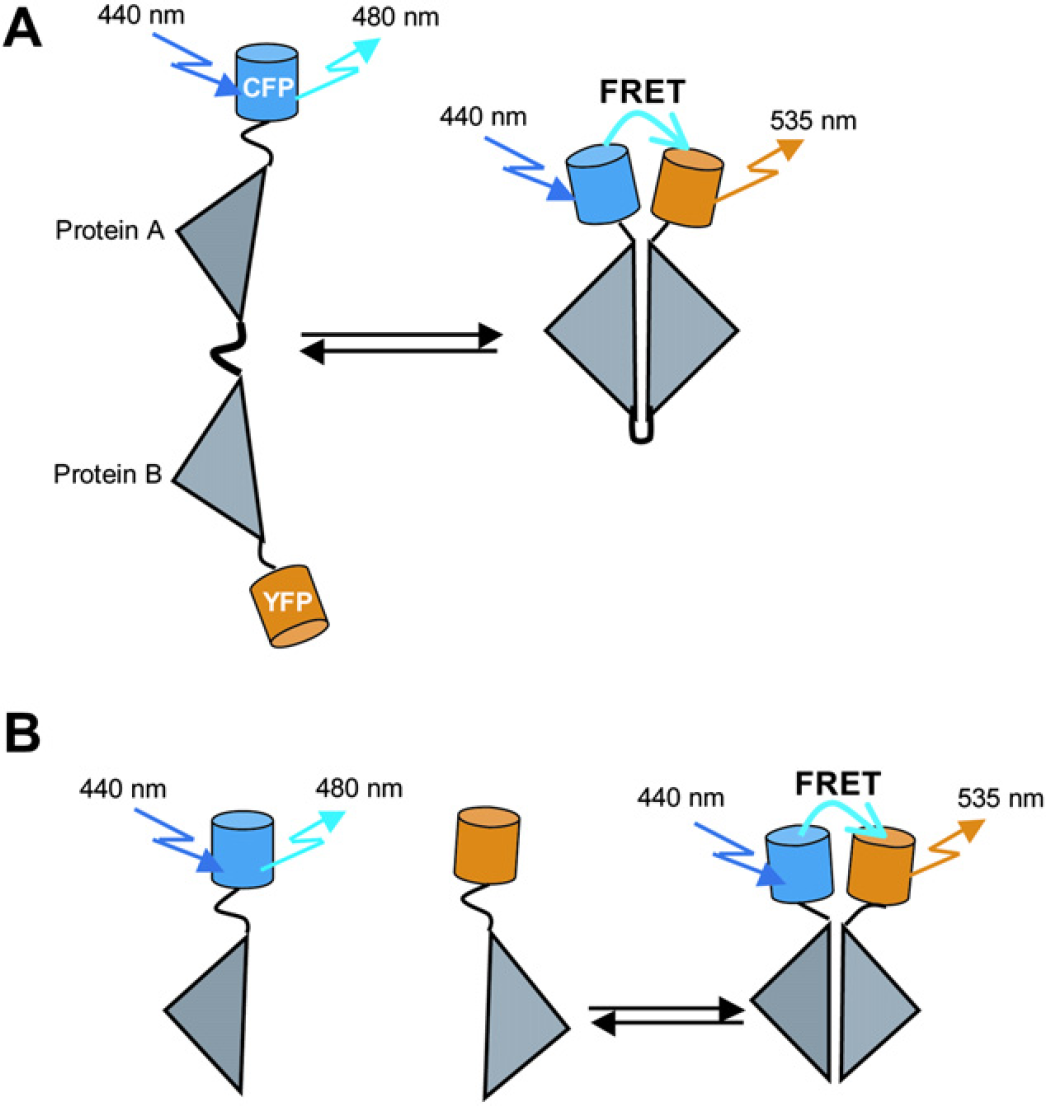
(A) Intermolecular FRET can occur between one protein (protein A) fused to the donor (cyan FP, CFP) and other protein (protein B) fused to an acceptor (yellow FP, YFP). When the two proteins bind to each other, FRET occurs. When they dissociate, FRET disappears. (B) Intramolecular FRET occurs when both the donor and acceptor fluorophores are on the same host protein.
Intramolecular fret probes for nuclear receptors
We will discuss and concentrate on intramolecular FRET probes to monitor intracellular process, especially the activation of nuclear receptors (NRs). Over the years, several researchers have developed intramolecular FRET probes using GFP variants for imaging intracellular Ca2+ concentration, 18 19 growth factor-induced activation of Ras and Rap1, intracellular chloride concentration in cultured hippocampal neurons, 20 cAMP, 21 cGMP, 22 signal transduction based on protein phosphorylation, 23 24 dynamics of AKT/protein kinase B, 25 histone acetylation, 26 phosphatase activities, 27 voltage-induced conformational changes in the voltage membrane-associated voltage-sensitive domain. 28
NRs are transcription factors and are important molecular targets for the development of drugs against different diseases. 29 31 We have developed intramolecular FRET-based probe molecules for imaging ligand-induced conformational changes in the NRs and their interaction with the coregulator and coactivator/corepressor 32 proteins in single living cells. These sensors exploit the large differential conformational changes of the helix12 in the NR ligand-binding domain (NR LBD) on binding a ligand of varying pharmacology. The LBD of all NRs has a common overall three-dimensional structure. 33 34 A ligand binding to the NR induces a conformational change in the NR LBD, which allows the ligand-bound NR to interact with coregulator proteins, coactivators, and corepressors. The coactivator binding to the NR results in the activation of gene expression related to the NR functions; however, corepressor binding to the NR suppresses the gene expression in the cell. Pure agonists force the LBD to adopt an active conformation, which generates a hydrophobic groove on the surface of the LBD to accommodate and recruit a coactivator to the NR, whereas a pure antagonist-induced conformational change inhibits the recruitment of coactivators to the NRs or recruit corepressor protein to the NRs. 35 39 However, in the case of selective NR modulators (SNRMs), an NR can recruit both coactivators and corepressors to NRs to stimulate or repress the NR transcriptional activity. The agonistic or antagonistic character of an SNRM depends on the expression levels of coactivator and corepressor proteins in a particular cell/tissue of the body. 40 41 The expression levels of coactivator and corepressor proteins are known to be very different between tissues. The dose of SNRMs thus results in the tissue-specific recruitment of coactivators or corepressors to NR. The molecular basis of the agonist, antagonist, and SNRM functions provides us with an idea for a rational method for high-throughput screening of NR ligands.
The principle of the optical probes is shown in Figures 2. The NR LBD is attached with a coregulator peptide and coactivator/corepressor via a flexible linker sequence. The resultant protein was inserted between cyan and yellow FPs (CFP, donor; and YFP, acceptor fluorophore, respectively) in such a way that excitation and emission spectra of CFP and YFP are suitable for FRET from CFP to YFP. This fusion protein functions as an optical probe for imaging ligand-induced conformational changes in the NR and their interaction with coregulator proteins in single living cells in an intramolecular FRET fashion. The addition of an agonist to cultured cells expressed with the fluorescent probe promotes interaction between the NR LBD and a coactivator peptide; this results in an increase in the FRET from CFP to YFP that was measured by a decrease in the CFP/YFP ratio. By contrast, an antagonist inhibits NR LBD/coactivator interaction within the fluorescent probe. The addition of an SNRM ligand that has mixed agonist/antagonist character promotes the NR LBD interaction with the coactivator and the interaction between the corepressor to increase the FRET response. The strategy was used to discriminate among NR agonists, antagonists, and SNRMs. We have developed fluorescent probes for progesterone, androgen, estrogen, glucocorticoid, and peroxisome proliferator-activated gamma receptors (PR, AR, ER, GR, and PPARγ, respectively). These probes were named CON-PRO (ligand-induced
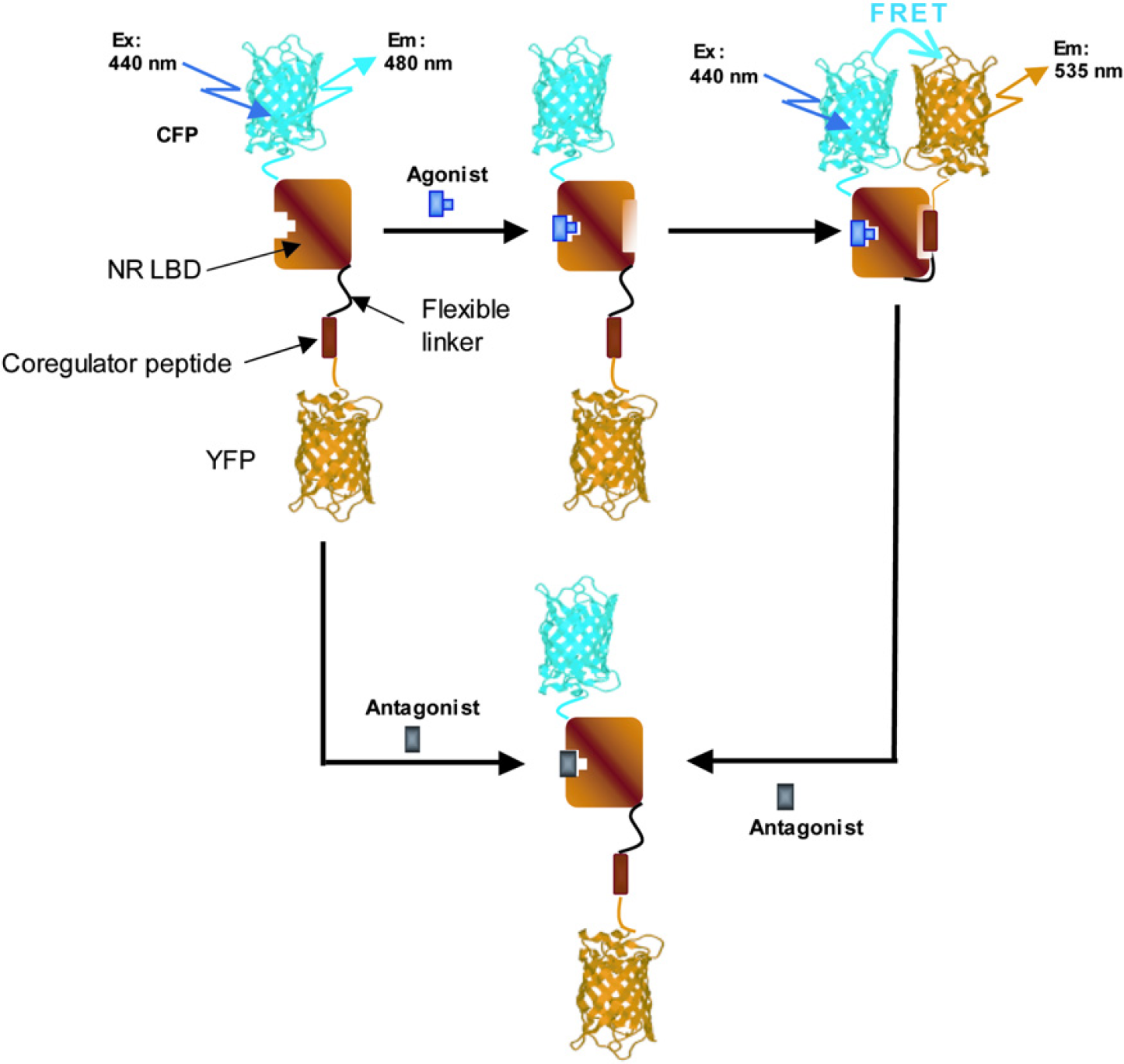
Principle of the intramolecular FRET probe to visualize the ligand-dependent interaction between the NR LBD and the NR-interacting coregulator peptide. An agonist promotes binding between the NR LBD and the coactivator. Consequently, CFP is oriented in close proximity to YFP, resulting in an increase in the FRET response. By contrast, an antagonist prevents binding of the coactivator peptide to the NR LBD. Replacement of an agonist with an antagonist results in dissociation of the receptor/coactivator complex to abolish the FRET change. Magnitude of the FRET increase strongly depends on the relative orientation and distance between the donor (CFP) and acceptor (YFP) fluorophores.
Imaging interaction between pr and coregulator proteins in real time using conpro
The response of the CONPRO was evaluated under a fluorescence microscope. Cultured cells were expressed with CONPRO and stimulated with progesterone (100 nM), an endogenous PR agonist. The emission ratio of CFP to YFP (CFP/YFP) was observed to decrease steadily and then level off after ∼ 10 min (Fig. 3A). To confirm that the increase in the FRET was actually triggered by progesterone binding to the PR LBD followed by the recruitment of co-activator to the PR, we made two CONPRO mutants, CONPRO/mutantLBD and CONPRO/3A. As expected, neither of these two mutants showed any significant change in the emission ratio of CFP/YFP on progesterone stimulation. Mifepristone (RU486), which is a selective modulator for the PR and shows mixed agonist/antagonist activity, 41 is used for contraception. 47 The response of the CONPRO was monitored by adding RU486 (100 nM) to cultured cells expressed with CONPRO. A large increase in the FRET signal was observed with RU486 similar to progesterone (Fig. 2B). This demonstrates the agonistic behavior of RU486.
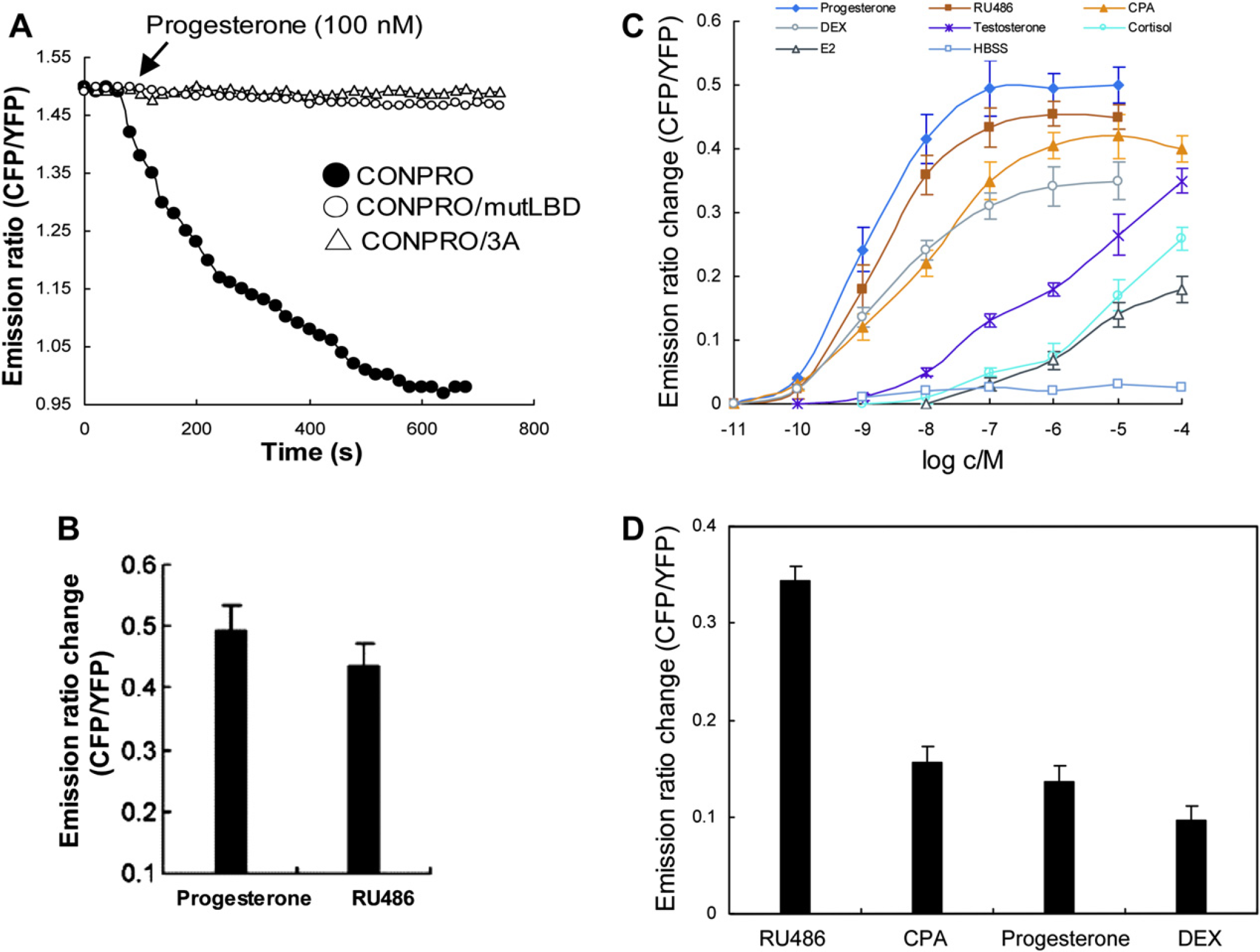
FRET was expressed as emission ratio of CFP to YFP signals. (A) Time course of the FRET responses on addition of progesterone to live PK-15 cells expressed with CONPRO (•), CONPRO/mutLBD (O), or CONPRO/3A (Δ). To construct CONPRO/mutLBD, the cysteine residue of the PR LBD necessary for the progesterone binding was replaced with the serine residue. To construct CONPRO/3A mutant, all the three leucine residues of the coactivator peptide that are necessary to interact with the PR LBD are replaced with the alanine residues. Each time course is one of five independent experiments. For each experiment, 0.8 μg of the expression vector encoding CONPRO, CONPRO/mutLBD, or CONPRO/3A was transfected to cultured cells in 3.5-cm glass-based dishes. A single cell was selected from each dish to monitor the effect of progesterone on CONPRO, CONPRO/mutLBD, or CONPRO/3A. (B) Changes in the CFP/YFP emission ratio of CONPRO on stimulation with progesterone (100 nM) and RU486 (100 nM) each 100 nM RU486 in PK-15 cells. The results are the means ± standard deviation (SD) of emission ratios from five different experiments. (C) FRET responses of CONPRO for various concentrations of different compounds. The results are the means ± SD of emission ratios of three cells from three different experiments. For each experiment, a single cell was imaged from a 3.5-cm glass-based dish to visualize the effect of each concentration of the tested ligand. (D) Emission ratio change on addition of 1.0 μM each RU486, CPA, progesterone, and DEX to cultured cells expressed with CONPRO1. The results are the means ± SD of emission ratios from three different cells in three experiments.
Figure 3C shows the dose-dependent increase in the FRET response with progesterone, RU486, and other steroidal compounds, such as cyproterone acetate (CPA, a synthetic progestin and antiprostate cancer drug), dexameth-asone (DEX, a PR-targeted drug), testosterone (a natural AR ligand), cortisol (a natural GR ligand), and 17β-estradiol (a natural ER ligand). The differences in the FRET observed by the tested ligands may be because of several factors, such as differences in ligand affinity for the PR LBD, differences in ligand ability to induce conformational change in the PR LBD to interact with the coactivator peptide, and differences in the rates of cellular influx/efflux of the ligands.
The compounds applied to CONPRO in Figure 3C were also applied to CONPRO1 (in CONPRO1, a corepressor peptide was used instead of coactivator) to evaluate their abilities to promote PR LBD/corepressor interactions. RU486 displayed maximum FRET response, CPA and progesterone showed significant response, and DEX elicited a weak FRET response as shown in Figure 4D. RU486 also showed both agonistic and antagonistic activities on GR. 45 The results demonstrate that an SNRM has the ability to induce recruitment of corepressor and coactivator proteins to an NR LBD depending on the availability/relative concentration of coactivator and corepressor proteins in a certain cell/tissue of the body to stimulate or block the transcriptional activities of the NR. 41
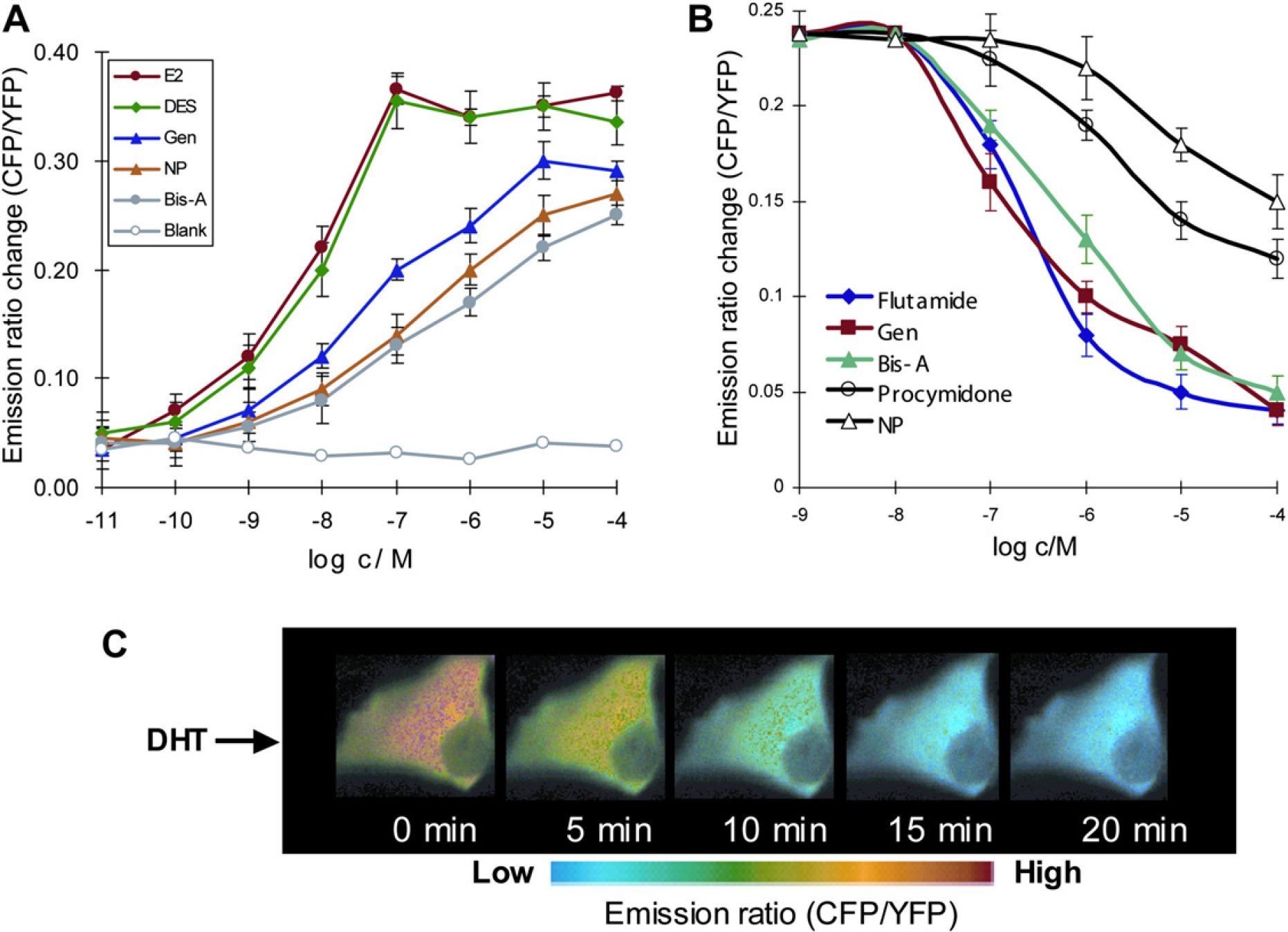
(A) Response of ER SCCOR probe for various concentrations of E2 and EDCs; diethylstilbestrol (DES), genistein (Gen), non-ylphenol (NP), and bisphenol A (Bis-A). The results are the mean ± standard deviation of emission ratio from five different cells/experiments. (B) Emission ratio change for DHT (10 nM) was assessed in the presence of various concentrations of flutamide, Gen, Bis-A, procymidone, and NP, respectively. For each experiment, the antagonist of various concentrations (1.0 nM—100 μM) was added to glass-base dishes containing PK-15 cells expressing the AR FICARO probe, and the resultant mixture incubated for 10-12 min at room temperature. Cells were imaged, and 10 nM of DHT was added in the same cells without washing the antagonist to monitor the inhibitory effect of the antagonist on the DHT-induced receptor/coactivator interaction within FICARO. The results are the means ± SD of emission ratios from three different cells/experiments.
There is mounting evidence that many synthetic chemicals having little structural resemblance to natural hormones mimic or block the natural hormone activities in the living body by binding with the steroid receptor. 47 51 These chemicals are also called endocrine-disrupting chemicals (EDCs). Early developmental exposure to such chemicals is known to cause reproductive tract abnormalities, decrease in reproductive organ weights, and low sperm counts and quality in wildlife and humans. 52 54 We used ER SCCOR, AR FICARO, and GR GLUCOCOR probes to evaluate the abilities of EDCs on activation or suppression of steroid receptors as shown in Figure 4A,B. Figure 4C shows pseudocolor images of cells expressed with AR FICARO probe when stimulated with dihydrotestoster-one (DHT), and it illustrates a DHT-induced change in the CFP/YFP emission ratio of the probe throughout the cell. Our genetically encoded FRET-based fluorescent probes are useful to identify any NR ligands that activate/inhibit the NR action through genomic pathway. We have also used these probes to show that ginsenoside Re, which acts as a specific agonist for the nongenomic pathway of sex steroid receptors, behaves like an antagonist by inhibiting the coactivator recruitment to the steroid receptor in the genomic pathway. 55
Protein-fragment complementation assay for ligand-induced nr conformational changes and protein interactions
Protein-fragment complementation (PCA) is another popular method to monitor the activities of small biomolecules, protein—protein interactions, and protein conformational changes. For the PCA, a reporter protein is rationally dissected into two fragments using protein engineering strategies, and protein—protein interactions are measured by fusing each of the protein of interest to two complementary fragments of the reporter protein. On association of the two interacting proteins of interest, the unfolded reporter protein fragments are brought into proximity. This allows the reporter fragments to complement and fold into the unique three-dimensional structure of the reporter protein and reconstitute its activity. 56 58 Using this strategy, inter-or intramolecular PCA-based biosensors can be designed. The reporter can be a fluorescent, such as GFP, or bioluminescent, such as luciferase, to develop a PCA. 59 60 The potential of the luminescent PCAs was explored to noninvasively monitor caspase activities, protein translocation, protein conformational changes, drug—protein interactions, and protein—protein interactions in living mice. 60 63
We have developed a reversible intramolecular PCA-based biosensor for the androgen receptor ligands, agonists, and antagonists. Firefly luciferase (FLuc) was dissected into N-terminal (1-415 amino acid [aa]) and C-terminal (416-550 aa) fragments. The AR LBD and the FQNLF motif of the AR N-terminal domain were inserted into the N-and the C-terminal of the luciferase fragments (FlucN and FLucC), respectively (Fig. 5A). This single molecule-format bioluminescence probe (SIMBI) was expressed in HeLa cells, and the cells were stimulated with various concentrations of different steroids and nonsteroids. 64 Stimulation of the cells expressed with the probe by DHT promoted association of the LBD and the N-terminal peptide within SIMBI, which resulted in a ∼ 50-fold increase in luminescence. The reversibility of the luciferase complementation in SIMBI was also demonstrated clearly in cultured cells. The DHT-induced luminescence intensity was decreased to baseline in ∼ 2 h, when the culture medium of the HeLa cells expressed with SIMBI stimulated with DHT was replaced with the fresh steroid-free medium (Fig. 5B).
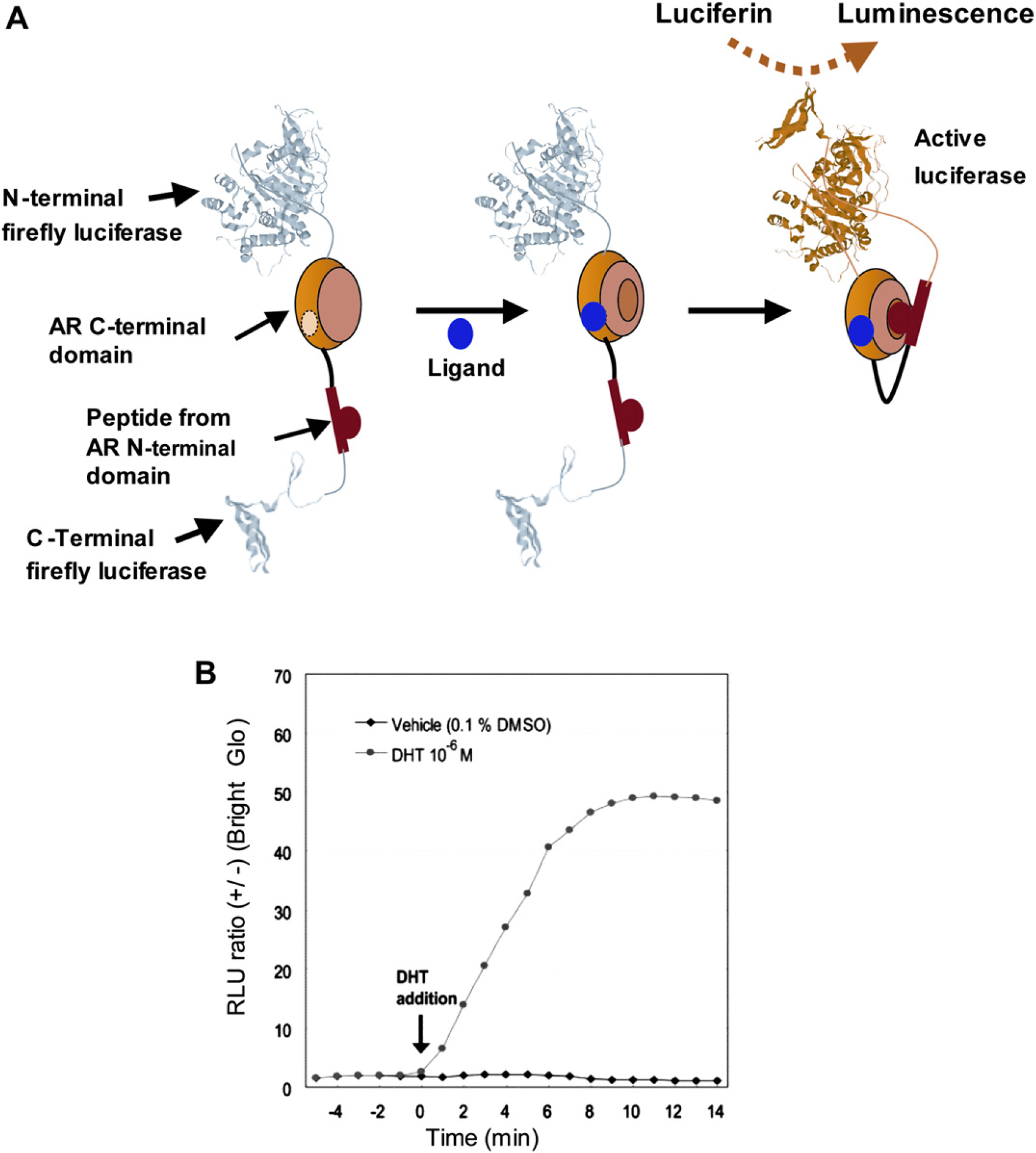
(A) Principle for the detection of intramolecular conformational change of AR. An androgen induces the conformational change of the AR LBD and its subsequent association with the FQNLF motif of the N-terminal domain of AR. The association triggers the recovery of the FLuc activities by the protein complementation of split N-and C-terminal fragments of FLuc, and the recovered luciferase activities were taken as a measure of the androgenicity of ligands. (B) Determination of the ligand-dependent kinetics in the luminescence intensities from SIMBI. HeLa cells carrying SIMBI were stimulated with DHT (1.0 μM) or vehicle (0.1% DMSO) at time 0, and the dynamics of the luminescence intensities were recorded for 14 min.
Recently, we engineered a new luciferase-fragments pair for the PCA; the reversibility of the PCA using these luciferase fragments was also shown by association and dissociation of FK506-binding protein (FKBP) and FKBP binding domain (FRB) complex in the presence of rapamycin (promoter of FKBP—FRB interaction) and FK-506 (inhibitor of FKBP—FRB interaction), respectively. In complicated protein interaction networks, a protein of interest might interact with proteins of many kinds in single cells. In such cases, it would be more useful if a common C-terminal fragment, which has the ability to complement multiple N-terminal luciferases, for the measurement of simultaneous interactions. To achieve this, we randomly mutagenized by site-directed mutagenesis to develop a mutant of C-terminal fragment from click beetle red (CBR) luciferase. This mutated C-terminal fragment was connected with FRB and the N-terminal fragments of FLuc, Emerald Luc (ELuc), or CBR were connected with FKBP. Among the mutant proteins that were screened, a mutant of the C-terminal fragment including three point mutations, F420I, G421A, E453S, named multiple-complement luciferase fragment (McLuc1), demonstrated the most remarkable properties, which enabled complementation to all N-terminal fragments of FLuc, CBR, and ELuc. Remarkably, absolute photon counts of a complement of McLuc1 and N-terminal FLuc exhibited a 12-fold increase compared with those of a native pair of N-terminal and C-terminal FLuc. The signal-to-background ratio on using McLuc1 was improved to 10 times higher than that of the native one. Use of McLuc1 with N-terminal ELuc demonstrated 40-fold stronger bioluminescence than that of native ELuc fragments. By using the combination of multicolor N-terminal fragments of click beetle luciferases with McLuc1, the discrete pairs of protein—protein interactions among Smad family members in the same living cells were shown with the help of green and red band-pass filters. The complementation of McLuc1 with the N-terminals of green and red luciferases has also been shown beautifully for imaging of protein interactions in single Xenopus embryo and living mice. 60
Conclusion
Fluorescence (FRET) and bioluminescence (PCA) assays are relatively simple, and there is no need for the production of recombinant proteins or protein purification, which is often a problem for the development of large scale in vitro assays for drug—protein interactions. The FRET and PCA assays are reversible and quicker to perform in single living cells. These assays can be performed to monitor interactions between more than two proteins simultaneously. The assays are not intended as a read-out of the binding affinity of a drug, but rather they probe the drug's efficacy as an agonist, antagonist, or SNRM in living cells. The permeability of a drug into cells and the conformational changes that are induced in a receptor to regulate the interaction with other intracellular proteins determine the efficacy of a drug much more than a simple binding assay. Therefore, the FRET and/or PCA assays can play a significant role in the development of drugs against different diseases.
Acknowledgments
Work reviewed herein was supported by the Japan Society for the Promotion of Science (JSPS), Japan Science and Technology Agency (JST), and New Energy Industrial Technology Development Organization (NEDO).
Competing Interests Statement: The author certifies that he has no relevant financial interests in this manuscript.