
Abstract
Increases in the use of protein-based pharmaceuticals require the development of cost-effective methods of storage and transport of sensitive biomolecules. In this article, we review the general problems of protein stabilization, aspects specific to antibodies, and a proposed method for protecting proteins based on nanostructured hydrogels. This review is not intended to be comprehensive, but instead to provide the reader with specific examples that capture some of the key challenges and opportunities of the field.
Introduction
Major strides have occurred in protein design and manufacture. For example, through site-directed mutagenesis, the amino acid sequence of a protein can be altered to beneficially increase activity, lower immune response, and increase stability. Additionally, histidine tails can be added to the terminus of a protein to simplify isolation via immobilized metal affinity chromatography, and thus lessen the contaminant background of the final isolate. It is even possible to minimize inclusion body formation within the cell that produces the protein, which simplifies the downstream isolation and purification processes. Finally, metabolic engineering has enhanced the yield of protein from recombinant cells.
The proteins that have emerged from biotechnology fall into different categories. Enzymes have long been produced by microbial or fungal processes, and used for nonmedical purposes. One example is provided by the protein-degrading enzymes (proteases) that improve the cleansing power of detergents. Now, enzymes such as streptokinase are produced by recombinant bacteria, and used to treat blood clots and myocardial infarction. Small and large proteins lacking enzymatic activity have also increasingly entered or passed clinical trials. For example, insulin produced by recombinant bacteria is now routinely used to manage diabetes, thereby supplanting the original porcine source. More recently, a host of antibodies have emerged as potential treatments for cancer, arthritis, and other chronic inflammatory and infectious diseases. About 200 such drugs are now in clinical trials, and a few are already on the market. 1
Despite these advances, all protein products still suffer from two classic problems. First, the tertiary structure is prone to unfolding at ambient or elevated temperatures. This and other events can also promote the dissociation of subunits in oligomeric proteins. 2 The resulting short shelf life increases the cost of inventorying and administering proteins. From a larger perspective, a less than perfect “cold chain” presents challenges in disseminating efficacious therapeutics to developing countries or deep within rural areas.
Secondly, proteins are prone to aggregation, which entails the “sticking together” of individual molecules driven by weak colloidal-level forces. Native structures or alterations of native structures can participate in aggregation reactions, which are also known as self-association. For example, under some solvent conditions α-chymotrypsin, which is ellipsoidal, can form linear end-to-end aggregates, which also influence transport properties. 3 Glutamate dehydrogenase, in turn, can undergo an “indefinite self-association,” resulting in aggregates that are unbounded in size. 4 Finally, prions represent an extreme example where refractory and deleterious protein-based deposits collect within the nervous system. Overall, the combination of unfolding and aggregation conspire to reduce the shelf life of a protein-based product.
To contend with aggregation, we have begun to explore another approach that may realize the following three design goals. First, the protein need not be reengineered to reduce aggregation. This may be advantageous because protein molecules are highly cooperative structures, and reengineering a molecule to have reduced aggregation may or may not also result in reduced activity and/or stability. Secondly, the concentration of the stabilized protein solution should be reasonably high to provide different dosage levels and low injection volumes. Moreover, stable storage at 10 to 100 mg/ mL could be envisioned for production stockpiles, where dilution could be done at the time of shipping or at the point of use. That would facilitate the inventorying of, for example, multiple versions of a vaccine, where one could attempt to remain ahead of likely mutations or possible pandemics. Third, the approach should potentially offer the possibility of increased stability against thermal unfolding.
A potential approach entails using ordered arrays of nano-porous structures. This article first reviews some facets of protein aggregation, the deleterious effects, and some current remedies. Although all protein types suffer from denaturation and aggregation, the initial focus will be on antibodies. This focus enables conciseness while also highlighting the issues through the example of a current, make-or-break therapeutic product class that is now being examined in many clinical trials. Then, an approach for protecting proteins using soft nanostructured hydrogels is described. Thereafter, some initial results using albumin as a protein model are described.
Protein Aggregation, Suppression, and Impact within the Context of Antibodies
To first provide some structural background, antibodies, which are also referred to as immunoglobins, consist of four chains. The region that binds the antigen is known as the Fab region, which is an abbreviation for fragment, antigen binding. The so-called fragment, crystallizable region (Fc) lies at the “stalk” of the “Y” shape. The Fc region can bind to specific proteins, thereby enabling each antibody to evoke a particular immune response for a given antigen. The Fc region also binds to different cell receptors and other immune molecules such as complement proteins. After binding, outcomes such as cell lysis or phagocytosis occur where the latter is enabled by the masking of the target's negative charge (i.e., opsonization). An Fc fragment is typically glycosylated.
A typical molecular weight is 150 kDa. Representative dimensions are 14.2 × 8.5 × 3.8 nm and the need to characterize these sorts of dimensions will be made clear later in this review. Based on a diffusion coefficient equal to 4.3 × 10−7 cm2/s, the equivalent hard sphere radius is 1.4 nm. Differential calorimetry scans typically exhibit multi ple peaks over 60 to 100 °C, where each peak corresponds to the endothermic unfolding of a particular domain. There are about 200 monoclonal antibodies in human clinical trials. 1 This number represents about 20% of therapeutic proteins.
Industrial practitioners have noted that a need exists to better define and control the nature of the protein product starting with the isolation step(s) and through storage and dissemination. 1 Indeed, whether aggregation results during purification or storage, the adverse side effects are numerous. The simplest outcome is that changes in biological activity occur, which complicates dosage or more critically, exposes the patient to adverse side effects while falling short of the therapeutic level of efficacy. The adverse side effects that can arise from infusing a patient with aggregated protein include anaphylaxis and organ failure. Because the side effects are serious and efficacy reduction complicates dosing, the only recourse is to conservatively place an expiration date on a product and then discard outdated products, which is yet another contribution to the expense of medical care.
A study conducted at MedImmune (Gaithersburg, MD) outlines some general factors that may play a role in aggregation. 5 When comparing along a family of antibody derivatives, if the Fc region is conserved, intermolecular interactions tend to be covalent and thus result in irreversible aggregation. The Fab region may play a dominant role in this case. These types of interactions are not responsive to the presence and types of excipients (solution additives). Mutations in the Fc region result in poor stability (i.e., heightened aggregation) and intermolecular interactions were suggested to be noncovalent and thus reversible. Sensitivity to excipients was, in turn, found to be high in this case.
Another study focused on aggregation in lyophilized preparations where excipients were present or absent. 6 Less aggregation was found to occur in lyophilized formulations of humanized monoclonal antibodies containing the carbohydrate excipients, sucrose and trehalose. The reduction of sulf-hydryl content indicated that aggregation, in part, occurred through the formation of intermolecular disulfide bonds. The chemical environment, as well as the conformational changes permitted in an anhydrous environment, were thought to account for the differences in aggregation. The basis for the efficacy of the carbohydrate excipients was noted to follow from the factors that control protein conformation. 6 First, water drives the so-called “hydrophobic effect” where tyrosine and other hydrophobic amino acid residues are driven to the interior of the folded protein structure. The entropic release of water also contributes to the folding process. Second, water also enables the formation of hydrogen and other polarity-based bonds, which are also stabilizing. In the absence of water, both stabilizing factors are altered, thereby potentially allowing a protein to adopt alternative and reactive structures. In contrast, when carbohydrate excipients are used, a protein finds itself in a glassy viscous environment that can limit conformational excursions. In tandem with protein dilution, the intermolecular encounters that lead to aggregation are kinetically suppressed owing to the high viscosity of the medium. Additionally, carbohydrates contain hydroxyl groups that may chemically substitute for water in polarity-based bonding. Of course, these same advantageous properties lead to slow reconstitution of the antibodies because of the high viscosity of the medium.
Another physical methodology involves total solvent removal and is commonly known as freeze drying or the alternative of spray drying. These techniques require excipients to hinder protein damage during processing. In many cases, a significant loss of viability is seen on reconstitution of proteins—in fact, some proteins simply cannot be treated in this way. However, long-term storage is cheap and successful for those proteins that are robust enough for treatment. Success of the latter approach requires the understanding of the weak forces that hold protein structure together and the tools that can be used to maintain these forces. Confinement,7-12 mac-romolecular crowding,13-20 and control of water structure 21 fall into this second class and offer the opportunity to protect more sensitive proteins. These approaches offer simpler reconstitution and less loss of viability, but they are not as well understood and are much more protein specific.
Another strategy under investigation is implemented at the protein sequence level. It entails using the computer analysis of protein structure to elucidate how to engineer a less aggregation-prone molecule. For example, one model dubbed SAP (spatial aggregation propensity), executes a dynamic, three-dimensional simulation of antibody molecules. 22 Unlike static representations such as those provided by x-ray crystallography, hydrophobic regions and their extent are determined when the molecule is in solution. Through molecular engineering, these regions where inter-molecular interactions can potentially occur through hydrophobic interactions may be “edited out” by, for example, changing the amino acids involved.
Overall, whereas aggregate formation can occur during protein isolation, the process also can occur during storage in solution. Excipient-based technology relies, in part, on the dilution of the antibody product. Constraining conformational changes and physically suppressing the rate of intermolecular interactions comprise the favorable outcomes provided following dilution. Effective doses of antibodies can actually be quite high; hence, dilution could be problematic. Carbohydrates are not usually regarded as toxic, but injection directly into the bloodstream could also elicit adverse side effects in some patients. Concerning molecular engineering, this approach may indeed be promising; however, biological molecules can be highly cooperative structures. Thus, accentuating or diminishing one property may affect other attributes in some cases. Thus, we have begun to explore the use of nanotechnology for achieving some of the outcomes provided by using excipients while lessening the need for protein product dilution. Other motivational facets exist as described next.
Motivation for Nanostructured Approach
As mentioned previously, there are many current protection methods: one is to completely remove the protein from solvent, whereas a second focuses on maintaining some of the structure driven by solvent. Here, we describe an approach to protein protection that takes advantage of both confinement and macromolecular crowding. Aqueous block copolymer solutions are allowed to self-assemble around target proteins to form nanoscale “pockets” to protect the protein. The process is reversible and differs from other uses of these polymeric materials in that the nanoscale structure formed is key to the mechanism.
Macromolecular crowding has been investigated as a method of protecting proteins based on the observation that physiological fluids inside cells contain concentrations as high as 30 to 40 vol% of macromolecules. 8 High concentrations of macromolecules that do not specifically bind to protein provide steric repulsions that affect protein folding in a densely packed fluid. Thermodynamic arguments based on changes in the chemical potential of the solvent have been proposed and crowding argued to be a relevant protection mechanism in living systems. Crowding agents such as Ficoll 70 (an inert polysaccharide) have been added to solutions of the globular protein apoflavodoxin and support the hypothesis that macromolecular crowding increases protein stability. As Ficoll 70 concentration is increased (from 0 to 400 mg/mL), the denaturation temperature increases from 45 to 65 °C. This stabilizing effect that Ficoll 70 had on the apoflavodoxin was also observed through far-UV circular di-chroism that showed maintenance of secondary structure as the concentration of the crowding agent increased. 18 This is one example of effective macromolecular crowding; polyethylene glycol (PEG), dextran, polysaccharides, and other “inert” polymers have been shown to protect proteins from different causes of denaturation (urea, temperature, pH, and so forth). 23
Molecular confinement is another approach to protein protection. This has been studied by encapsulating proteins within the pores of a silica glass. The circular dichroism spectra of lysozyme, α-lactalbumin, and metmyoglobin were similar in buffer solution and inside the silica matrix indicating that the encapsulation procedure does not denature the proteins. The encapsulated proteins all showed an improved resistance to aggregation caused by water-miscible alcohols (hexafluoroisopropanol and ethanol). The protein stability of the encapsulated proteins was also tested thermally up to 95 °C. Lysozyme and metmyoglobin showed irreversible denaturation in the silica matrix, but more of the secondary structure was maintained than in free solution. Also, the thermal denaturation of the encapsulated α-lactalbumin was completely reversible. This molecular confinement strategy was able to provide a 25 to 30 °C increase in the thermal stability of α-lactalbumin. 8 Denaturation of cytochrome c (hydrodynamic diameter ∼3 nm) encapsulated in a silica matrix with pore sizes of 2 to 6 nm was monitored by fluorescence spectroscopy in buffer solutions and solutions with high concentrations of the denaturing agent urea. Cytochrome c molecules that were encapsulated in larger pores demonstrated a higher level of unfolding than those encapsulated in smaller pores, indicating the need to match the nanometer length scales of pore to the specific protein. 20 One drawback of using the sol-gel method of protein encapsulation is that the interactions between the walls of the silica pores and the protein molecules can also have an effect on the stability of the protein and cannot be separated from the molecular confinement effect on protein stability. 20 Clearly, sol-gel grown silica does not provide a robust storage mechanism, as recovery of the protein at point of use is difficult (relying on diffusion out of the gel), but this work does demonstrate the importance of confinement in protein protection.
Templating Proteins in Nanostructured Hydrogels: Attributes and Parameters
We have been working for several years on the simple idea that the hydrogels formed by the packing of block copolymer micelles can be used to spatially template nanoparticulate material. We argue that nanoparticles will be sterically guided into the interstitial, water-filled, spaces between the micelles during a thermoreversible gelation. This simple approach was shown to be feasible using a combination of rhe-ology and contrast-variation neutron scattering.24-28 In those works, we demonstrated that nanoparticulate material will be templated into the interstitial spaces in a nanostructured hydrogel. We are investigating the applicability of this approach to protein storage and protection; hence, we will first describe the templating.
In general, amphiphilic block copolymers will form self-assembled structures when dispersed in solvents that are selective to some subset of the blocks. 29 Block copolymers are synthetic macromolecules with a sequence or “block” of one monomer connected to a “block” of a chemically different monomer. The geometry and long-range order of these structures depends on the chemistry of the polymer and the solution conditions among other controllable parameters.30-32 Long-range order in these systems occurs when the number of micellar aggregates (spherical, cylindrical, or planar) exceeds the crystallization volume fraction. The repulsion between monodisperse spherical micelles drives the formation of cubic phases (face-centered cubic or body-centered cubic) with order that persists over macroscopic length scales.29, 31, 33-37 Since the observation of these phases, they have been considered for use as templates to impart order to materials that lack this ability to self-associate and organize. Examples of methods to template discrete and continuous structures have been described.38, 39 Our recent work demonstrated the ability to organize discrete nanoparticles in thermoreversible block copolymer cubic crystals, an approach not used before our work.
Hydrogels prepared from concentrated solutions of triblock copolymers of polyethylene oxide (PEO) and polypropylene oxide (PPO), commercially known as Pluronic or Poloxamer, have been extensively studied over the past decade.31, 34, 36, 37, 40 The structure of the triblock is two blocks of PEO connected to a central block of PPO. All blocks of these copolymers are water soluble at low temperatures (< 5 °C) and very concentrated fluid dispersions are easily created. When the temperature is raised to values near room temperature, the middle PPO block becomes increasingly dehydrated and the polymers self-associate into micellar aggregates. At large polymer concentrations, the spherical micelles pack into micelle cubic crystals and the viscosity of the samples increases by more than four orders of magnitude. This transition is a gelation that occurs owing to the formation of micelles at a concentration high enough for them to pack into ordered arrays. This transition is fully reversible on repeated heating and cooling.
The thermoreversible ordering behavior of these polymers and their water solubility make them ideal template materials for the incorporation of nanometer-sized particulate additives (for example, proteins). We have used these templates to incorporate silica and gold nanoparticles as well as globular proteins.24-28 The approach is depicted in Figure 1. The particles are incorporated at low temperatures while the matrix viscosity is low and the block copolymer phase is disordered. Typical block copolymer concentrations are on the order of 20 to 30 wt% providing a solution that is primarily an aqueous environment for dispersion. Repulsive forces between the nanoparticles are crucial to achieve a stable dispersion in this concentrated polymer solution. The temperature is then raised and the ordered cubic gel is formed around the dispersed particles (proteins). The particles (proteins) remain trapped in the interstitial cavities of the cubic crystal and form an ordered array because these cavities also follow the crystal lattice.
Schematic of the approach. At temperatures below the gel temperature (typically 10—20 °C), the block copolymer is soluble in water and a dispersion of protein (•) and copolymer is created. As the dispersion is heated, the copolymers form micelles that spontaneously form a nanostructured hydrogel. The protein “goes along for the ride” and is sequestered into the interstitial spaces in the hydrogel. This process is reversible and cooling and/or dilution results in a dispersion of protein in the polymer, which is approved by the Food and Drug Administration.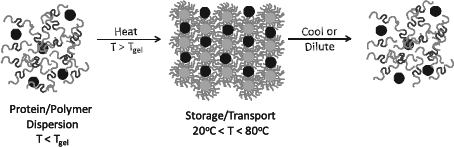
We have shown that much of the initial design can be based on the large volume of knowledge associated with atomic and hard-sphere colloidal crystals. One of the block copolymers that we have used forms a face-centered cubic (FCC) arrangement of the micelles in space. 31 Therefore, the crystal structure of these nanocomposites is expected to be analogous to the NaCl crystal. This crystal structure consists of an FCC crystal of smaller atoms (particles) intercalated in an FCC crystal of larger atoms (micelles). Although the stability of this phase in binary hard-sphere mixtures has been predicted by simulations and theory,41, 42 it was only recently observed experimentally in colloidal systems.43, 44 Although other binary crystal superlattices (ZnS and CsCl) could also occur, the theory of hard-sphere mixtures predicts a higher stability for the NaCl superlattice. 41 Our results are consistent with this picture and indicate that these swollen hydrogels structure in ways similar to colloidal crystals and atomic crystals. Although it is surprising that hard-sphere systems can provide suitable analogs for “soft” systems like block copolymer micelles, the agreement is likely because the primary driving force for organization is steric.
A major factor is the environment in the interstitial space in which the particle (protein) resides. The formation of the block copolymer crystal buries the hydrophobic portions of the block copolymer in the core of the micelle, protected from the aqueous phase by a dense corona of PEO. As the micelles pack into a crystal, the interstitial spaces, or “pockets,” are water filled with boundaries formed by the micelle corona. The length scale of the pocket is set by the crystal packing and micelle dimensions, which is tens of nanometers. So the “pockets” in the crystal are water-filled spaces with a high concentration of PEO bounded by even more concentrated PEO (the portion of the chain pinned to the core of the micelle). Because of the packing of the crystal, “hopping” between interstitial spaces is minimized, 45 resulting in limited transport of particles between the “pockets.” The “pocket” is a self-assembled, confined, water-filled space with length scales that can be matched to many proteins of reasonable molecular weight.
We hypothesize that this “pocket” provides a protective environment for proteins, which is based on three lines of reasoning. First, the “pockets” formed in nanostructured hy-drogels (Pluronic gels) provide confinement, which constrains conformational excursions. Second, macromolecular crowding can occur in an aqueous environment filled with concentrated PEO. Last, the confinement and crowding limits protein-protein interactions.
Most of our previous work has focused on one specific block copolymer, Pluronic F127, which forms spherical micelles that pack into an FCC crystal. We use this model system in our work because its solution behavior has been characterized extensively and the phase diagram is dominated by a single crystal structure.31, 34, 36, 37 The gelation temperature is near 15 °C and the solutions form stiff gels at room temperature and above. Contrast variation small-angle neutron scattering (SANS) was used to verify the templating.
Figure 2 shows typical data from the experiments. Circularly averaged SANS is shown as scattered intensity as a function of scattering angle, q. The peaks in these curves provide a fingerprint of a specific crystal structure. Researchers used to seeing small-angle x-ray scattering (SAXS) or crystallography results will note the breadth of these peaks, this is because of the low wavelength resolution of SANS—this disadvantage is offset by the ability to perform contrast variation experiments for aqueous samples. The two curves are from gels of the same composition, but different ratios of D2O:H2O in the solvent. The two different deuteration levels of the solvent render either the block copolymer or the dispersed phase (bovine serum albumin [BSA] in this case) invisible to the scattering. From the figure, we note that the peaks are at the same q position, indicating that the BSA dispersed in the gel has the same structure as the gel itself, which is consistent with particles dispersed in the octahedral sites of an FCC crystal.
Contrast-variation SANS from a 25 wt% FI27 matrix with 3% BSA dispersed. Scattering from the matrix (•) and BSA (O) are shown together. The coincidence of the peaks at 0.04Å−1 demonstrates that the BSA is templated and not randomly interspersed in the gel.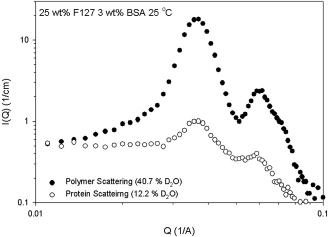
Our previous work used a combination of SANS and mechanical testing (rheology) to map out the operating space of the templating. These results elucidated the key parameters to consider. The first parameter is the proximity to the gelation temperature. The templating is optimal at temperatures more than 10 °C above the gelation temperature. Very high temperatures will also start to lose the templating as thermal energy in the nanoparticles provides enough energy to disrupt the nanostructure of the gel. For 25 wt% Pluronic F127 this provides an operating range of 25 to 85 °C. The second parameter is the loading of the gel. Forcing more than one particle per interstitial site will reduce the mechanical stability of the gel. Simple calculations of loading levels based on geometry can be used to estimate the maximum loading. For 25 wt% Pluronic F127, the optimal loading of BSA is equivalent to 3 wt%, or 30 mg/mL. The last parameter identified was the ratio, particle size/micelle dimension. This intuitively follows from the need for the nanoparticle to fit into the interstitial spaces of the block copolymer crystal. Through analogy to alloy and colloidal crystal literature, estimates of the optimal ratio of nanoparticle to micelle can be made. For Pluronic F127, the dispersed nanoparticles should be 9 nm in diameter or smaller, which accommodates antibodies as well as a host of other proteins.
We have demonstrated that we are able to template globular proteins in a nanostructured hydrogel.25, 26, 28 BSA and lysozyme (LYS) have been dispersed in different hydrogels. The loading of protein in the hydrogel is quite high, the equivalent of 30 mg/mL of protein in total volume. We have shown using contrast-variation SANS that the proteins are templated into the interstitial spaces in both cubically packed spherical micelle gels and hexagonally packed cylindrical micelle gels.25, 26, 28 We have observed that this templating method is quite general and can be applied to many particle systems as long as they can be initially dispersed in the concentrated polymer solution.
The requirements of the dispersed phase are not overly restrictive when considering proteins as the nanoparticulate material. Nanoparticles that can be dispersed in an aqueous solution of Pluronic are necessary. The particles do need to form stable dispersion, so repulsive interactions between particles are advisable, whether via hard sphere interactions or electrostatic repulsions, but both are common in protein solutions. Osmotic stresses in concentrated polymer solutions can be significant and electrostatic and/or hydration repulsive forces are essential to maintain particle stability. Concentrated polymer solutions like these would likely lead to depletion attraction in dispersions of larger colloidal particles; however, nanoparticles tend to have a higher repulsive peak in the interaction potential and depletion attraction is hindered. In fact, we have observed enhanced stability of the Pluronic/protein solutions as we approach the gel temperature, most likely because of the preference of the Pluronic unimers to form micelles over interaction with the proteins. 46
Significant Pluronic adsorption onto particle surface would lower the effective bulk concentration and likely disrupt the gel formation. In our early work, we used silica and gold nanoparticles based on the observation that adsorption, which would be strong on planar surfaces of the same material, is weak because of the high curvature and limited amount of surface area presented by nanoparticles. This assumption was primarily tested by measuring the critical micelle temperature (CMT), in the presence of increasing amounts of nanoparticle material. Peaks observed because of the endothermic micelle formation were found to occur at the same temperature indicating that the CMT is not significantly shifted as the particle concentration is changed from 1 to 10 wt%. These results indicate that any adsorption of polymer that may occur at the silica interface does not significantly alter the concentration of polymer in the bulk. Previous work indicates that adsorption of Pluronics occurs at the surface of silica particles, but the adsorbed amount at surface saturation is quite low (0.2∼0.4 mg/m2).47, 48 Specific adsorption of Pluronic onto the surfaces of proteins will need to be considered in the design of systems and strongly interacting systems avoided. However, many globular proteins, because of the presentation of charged, hydrophilic groups on the surface, do not exhibit adsorption of PEO or PPO (the components of Pluronic) on surfaces. 49
Once templated, the gels can be held at room temperature for long periods of time; note that the protein is trapped in a water-filled pocket bounded by PEO and only slightly larger than the protein itself. To validate the structural stability of the proteins within the templated gel, we either “melt” the gel back to its unimer state by cooling to below Tgel (10—15 °C depending on the block copolymer structure) or dilute the gel to a concentration low enough to inhibit micelle formation.
Initial Model System Results Showing Hindered Protein Aggregation
To begin verification of the proposed process, we chose to study BSA in a 25 wt% F127 Pluronic template. From earlier results, we know that this protein is templated into the pockets, but it is unlikely that the pocket is tight enough to protect the protein from thermal denaturation. That is, the Pluronic F127 at a concentration of 25 wt% has a “pocket” size of roughly 9 nm, which is larger than a BSA molecule, but close enough to template, or “trap” the proteins. However, this is an appropriate test case to verify that BSA can indeed be dissolved in the cold Pluronic, heated through the gelation (but not above room temperature), and then redispersed without significant colloidal aggregation of the relatively concentrated protein solutions. Solutions of 30 mg/mL BSA in deionized water and in deionized water and 25 wt% Pluronic were prepared, heated to room temperature, held for one week and then diluted back to 1 mg/mL BSA. The diluted solutions were characterized using dynamic light scattering (DLS) to determine the size distribution of protein in solution.
The results are shown in Figure 3. The dashed line indicates the size distribution of BSA held in concentrated, unbuffered, solution at room temperature. Most of the proteins are seen tightly dispersed around a hydrodynamic diameter of 4 nm and some aggregation is observed (larger aggregates near one micron in size), consistent with work seen in the literature.
50
To compare, the dotted line shows the size distribution of a solution of the same BSA stored at room temperature for one week at 1 mg/mL. Again, a large fraction of the BSA exists as monomers with some larger aggregates observed. The solid line indicates the size distribution of BSA that was concentrated in Pluronic and then templated into the interstitial spaces of the hydrogel. This was held at room temperature for one week and then diluted for size analysis. The BSA and Pluronic are diluted to the equivalent of 1 mg/mL BSA, so DLS is performed on a solution that contains BSA and just over 1 wt% Pluronic. Because a 1 wt% Pluronic solution is twice as viscous as water, the exact viscosity is measured and included in the DLS analysis (conversion of diffusivity to size). The key result found is the size remains tightly distributed and is likely still arising from monomeric BSA. Forcing the BSA into a structured gel as depicted in Figure 1 does not cause aggregation of the BSA. Interestingly, the largest part of the distribution observed in the system concentrated in water is not observed in the sample that was held in hydrogel. In fact, we have seen some evidence of the protein dispersion improving near Tgel when compared with aqueous solutions.
Impact of dispersing BSA in a nanostructured hydrogel compared to free in solution. DLS of diluted solutions that have been held for one week in different conditions: at I mg/mL in water (….), concentrated to 30 mg/mL in water (- - -), and concentrated to 30 mg/mL in a nanostructured hydrogel (—) show minimal change in size distribution once diluted back to I mg/mL.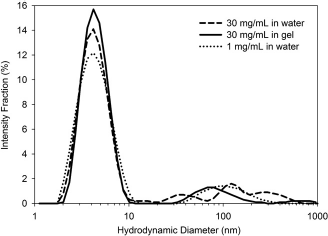
Overall, this preliminary work is encouraging. We find that the process of gelation and structuring does not induce significant aggregation of the BSA. These results provide impetus to investigate other proteins and a range of “storage” time scales and to investigate whether the nanostructured gels have the potential to protect proteins against thermal denaturation.
Conclusions
The increasing use of protein-based therapies requires that improved engineered solutions be developed that address the problem of storing these molecules such that denaturation and aggregation are suppressed. The article has described one strategy, which entails templating proteins in nanostructured hydrogels. The combination of molecular crowding and spatial confinement underlies the anticipated effectiveness of the approach.
The results obtained to date indicate that the concept works for the model protein, albumin, in that the process of templating the protein in a concentrated block copolymer solution as micellazation occurs does not cause significant aggregation of the protein. Also, over a relatively short one week time frame, the templated protein showed similar protection from aggregation as compared with a control solution (concentrated protein in water). The ranges of processing temperatures are mild, and significant protein loadings can be achieved that are of the order of tens of mg/mL. Temperature alteration or dilution can be used to “release” the protein. Moreover, the size requirement for the effective templating of a protein molecule excludes few proteins, and includes antibodies. To further test the strategy, other proteins will need to be examined as well as different storage times and temperatures.
Footnotes
Acknowledgments
Theresa LaFollette performed the protein aggregation studies and analysis; she acknowledges an NSF Graduate Fellowship for funding of her thesis work.
Competing Interests Statement: The authors declare that they have no competing interests.