
Abstract
UCB Selected Lymphocyte Antibody Method (SLAM) is a rapid and efficient process for the generation of high-quality monoclonal antibodies, in which variable region gene sequences are recovered directly from specific, single B cells. Monoclonal antibody generation has been limited in the past by the relatively low efficiency of the hybridoma process. UCB SLAM process is well suited to high-throughput screening and has been extensively automated at UCB. If necessary, in excess of 1 times 10 9 B cells can be screened in a campaign, to discover a rare therapeutic antibody candidate, which meets the stringent selection criteria. Primary screening for antigen binders, on purified or cell expressed antigen, is performed using a homogeneous fluorescence assay format. Supernatants from positive wells are consolidated to allow further secondary screening and selection of antibodies with desired characteristics. Individual, specific B cells are identified using a fluorescence based method and isolated using a micromanipulator. The antibody variable region genes are cloned from DNA extracted from the single B cell. The genes are sequenced then prepared for transient expression to confirm activity. Antibodies with affinities (K D) in the sub 10 pM range against a range of therapeutic targets are routinely recovered using this process.
Introduction
The original Selected Lymphocyte Antibody Method (SLAM) technology was in-licensed from Abgenix. 1 UCB SLAM is a further development of this technology, which has turned it into the cornerstone of our antibody discovery platform. It allows the selection of high-affinity antibodies from a broad pool of B cells for each new target. Within UCB's laboratories, this technology is operated in a high throughput and automated manner enabling antibody selection to be done on a scale not previously possible with hybridoma. The technology is applied to screening for therapeutic antibody candidates and for generating anti-mouse, rat, and rabbit research reagents. The UCB SLAM process has advantages when compared with hybridoma in that it increases the probability of discovering antibodies with desired characteristics, shown in Table 1.
Requirements for a therapeutic antibody

Because of the large number of B cells that can be screened using UCB SLAM, our criteria for selection of a therapeutic candidate can be extremely stringent. Throughout the development of UCB SLAM, we have used techniques and methods already being applied in high-throughput screening of chemical libraries, however automation and informatics challenges are different from those seen with chemical libraries. Handling of the cell culture is done aseptically requiring all liquid-handling robots to be installed within class II safety cabinets. Although the number of primary screen plates is relatively low, when compared with chemical library screening, in effect a new library of B cells is generated for each screening experiment. The number of B cells screened in each campaign can be 100 times greater than the number of compounds in a traditional chemical library.
Antibody Screening Method
A diagram showing the work flow of the UCB SLAM process is in Figure 1.
Primary screening of the cell culture supernatant is carried out using a homogeneous fluorescence assay in black clear-bottomed 384-well plates. This is a simple antigen binding assay and requires the sampling of supernatant from each cell culture well. A Thermo Scientific Matrix PlateMate Plus (Thermo Scientific, Hudson, NH) is used to transfer 10 μL of supernatant from the cell culture plate to the assay plate, in the process capturing the barcodes from both plates. The barcode information is saved as a text file and is automatically uploaded into the LIMS. A Thermo Scientific Multidrop (Thermo Scientific, Vartaa, FL) is used to add a fluorescently labeled anti-IgG Fc antibody and target antigen to the assay plates; the total assay volume is 30 μL. The target antigen can be in the form of either antigen labeled 10 μm polystyrene beads or antigen expressing cells; this type of assay requires no wash step; the assay plates are incubated for 1 hour before being read on an Applied Biosystems 8200 Cellular Detection System, (Applied Biosystems, Foster City, CA) with an integrated bar code reader and a plate stack linked to the reader via a Zymark Twister arm. Where antibody, conjugate, and antigen combine together, fluoresce will be associated with the bead or cell. This is detected as an image from a 1 mm 2 area of each assay well by the instrument software, and a hit is recorded; this is shown in Fig. 2.
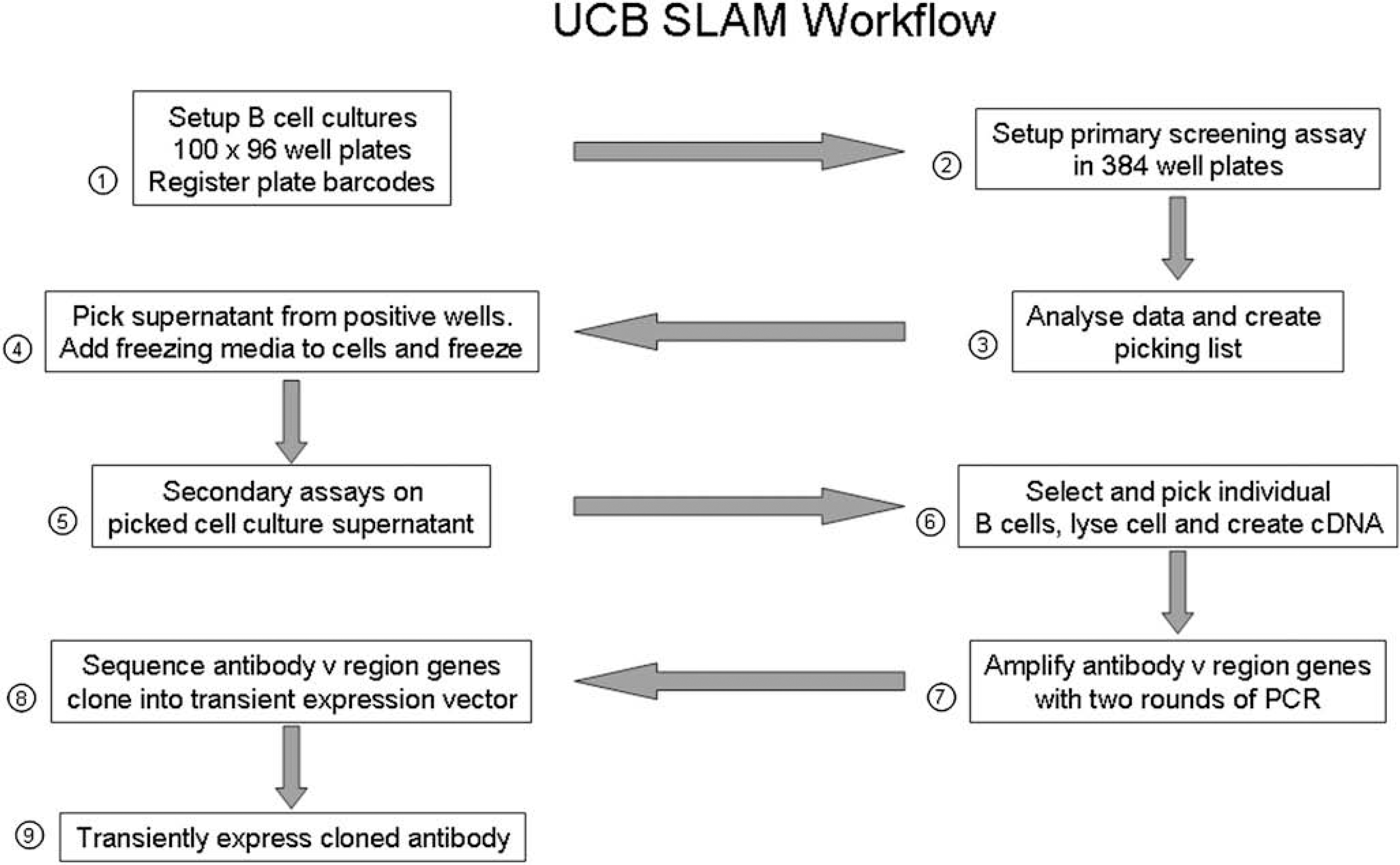
Work flow of UCB Selected Lymphocyte Antibody Method.
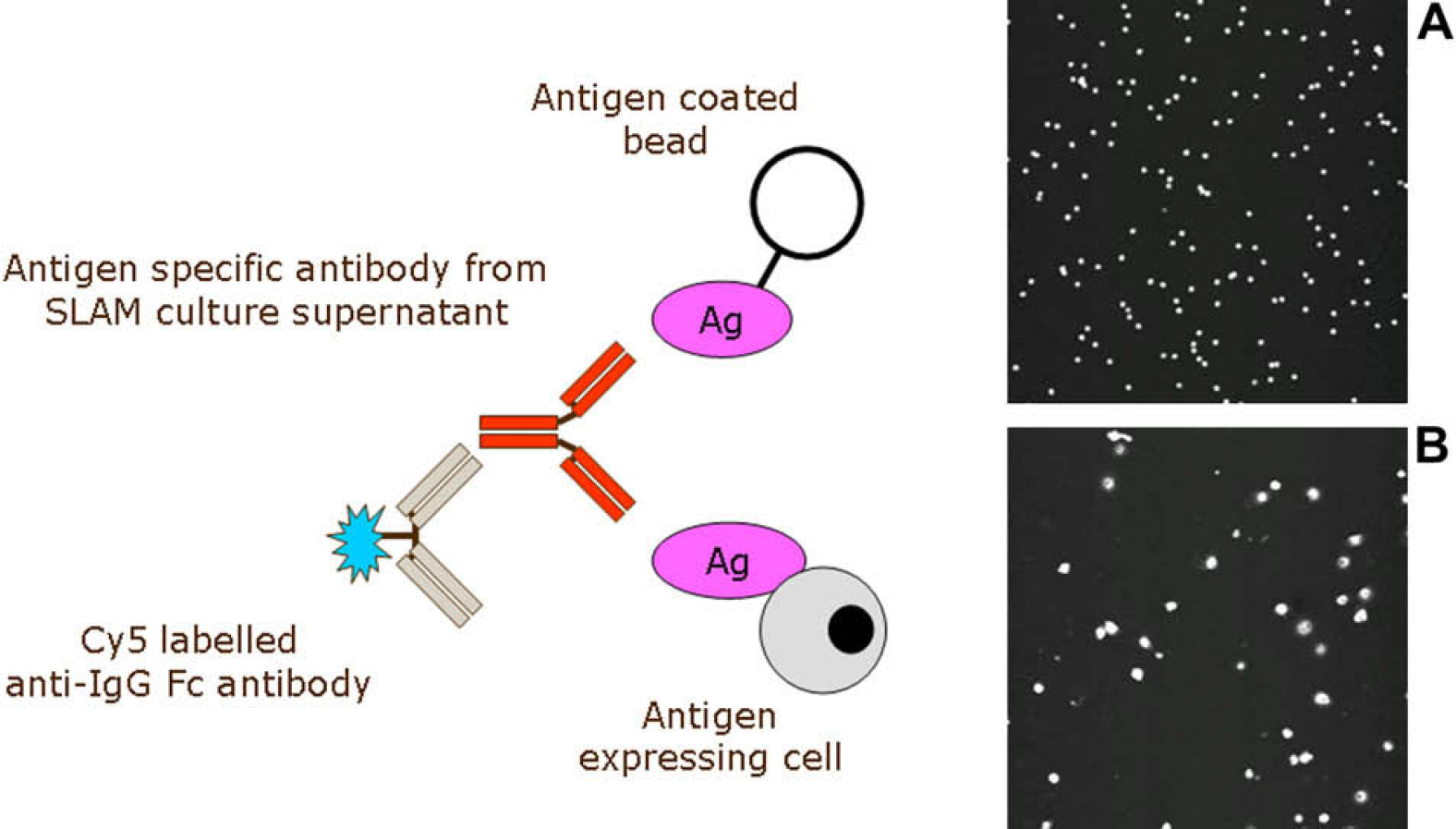
Homogeneous primary screening assay. Supernatant from the Selected Lymphocyte Antibody Method culture plates is mixed with either antigen coated beads (A) or antigen expressing cells (B), together with a Cy5 labeled anti-IgG Fc antibody. The 8200 Cellular Detection System scans each well with a 635 nm laser. Where specific antibody, conjugate, and antigen come together, the beads or cells fluoresce and a hit are recorded by the imaging software. The images A and B are of a 1 mm2 × 0.1 mm deep field from the center of the assay well, taken from the 8200 Cellular Detection System.
Culture Setup
B cells from immunized animals can be harvested from peripheral blood, spleen, bone marrow, and lymph nodes. Cells are added to cell culture media containing irradiated EL4 B5 feeder cells and T cell conditioned media, 1 before plating into 96-well cell-culture plates using a Thermo Scientific Matrix WellMate (Thermo Scientific, Hudson, NH) dispenser/stacker. Each experiment consists of 100 96-well plates; for each target, the number of experiments run in a screening campaign can vary. In excess of 25 experiments have been run. The number of cells added to each well can be adjusted from 500 to 10,000 in 200 μL of media to obtain a preferred hit rate in the primary screen. A hit rate of no greater than 10% of the wells showing positive, increases the probability of clonality when individual B cells are isolated from positive wells. Cells are cultured for between 5 and 7 days, during this time the B cells divide and secrete antibody into the media. The project and sample information, together with the cell culture plate barcodes are registered into a laboratory information management system (LIMS) to allow plate and sample tracking.
Assay Setup
Data Acquisition and Analysis
Primary screening data from the 8200 Cellular Detection System and plate barcode information from the Thermo PlateMate are uploaded into Activity Base (IDBS, Guilford, United Kingdom) together with assay parameters. Primary screening data from each experiment are analyzed in Spotfire DecisionSite (TIBCO, Palo Alto, CA), to select suitable wells to progress through to secondary screening. Where the hit rate is high, only a proportion of the positive wells, those with the highest fluorescent signals, are selected for progression.
Hit Picking
From each primary screen, the supernatants from positive wells are consolidated on master plates. The number of master plates picked can be between 2 and 10 depending on the hit rate. The Spotfire Decisionsite software produces a picking list of cell culture plate barcodes and well identifiers. The file containing the picking list is downloaded to an Aviso Onyx (Aviso Mechatronic Systems, Jena, Germany) liquid-handling robot, which transfers supernatants from the positive wells on cell culture plates over to a master plate. During this process, the samples are tracked through the Onyx software using the plate barcodes. After removal of the majority of the supernatant, freezing media is added, and the plates are slowly frozen and stored at −80 °C to preserve the B cells. The cell culture supernatant is then used in secondary screening assays.
Secondary Screening
Secondary screening may involve several assays to look at affinity, functional activity, site of action, and cross reactivity. These secondary screens vary greatly depending on the target antigen and are a mixture of cell- or protein-based assays. In addition, a Biacore A100 (Biacore Life Sciences, Uppsala, Sweden) has enabled the determination of the dissociation rate constants of hundreds of samples to be done rapidly. Data from these secondary screens are used to generate a list of candidates for single B cell identification and isolation.
Single B Cell Identification and Isolation
Selected wells will contain both antigen specific and antigen nonspecific B cells. To distinguish the specific from the nonspecific, cell-culture plates are removed from the −80 °C freezer, and selected wells are thawed and all the cells harvested. The well contents are incubated in cell culture media on a microscope slide at 37 °C, with antigen conjugated beads or cells expressing antigen together with a fluorescently labeled anti-IgG Fc antibody. During the incubation, the B cells will secrete antibody. A halo of fluorescence is seen in the immediate vicinity of the B cells secreting antigen specific antibody (Fig. 3). A fluorescent microscope (Olympus, Tokyo, Japan) with an Eppendorf TransferMan NK micromanipulator (Eppendorf Hamburg, Germany) is used to pick, single, antigen specific B cells and place them individually into a PCR tube. Several cells are selected from each well and placed in separate PCR tubes to ensure the correct antibody is isolated.
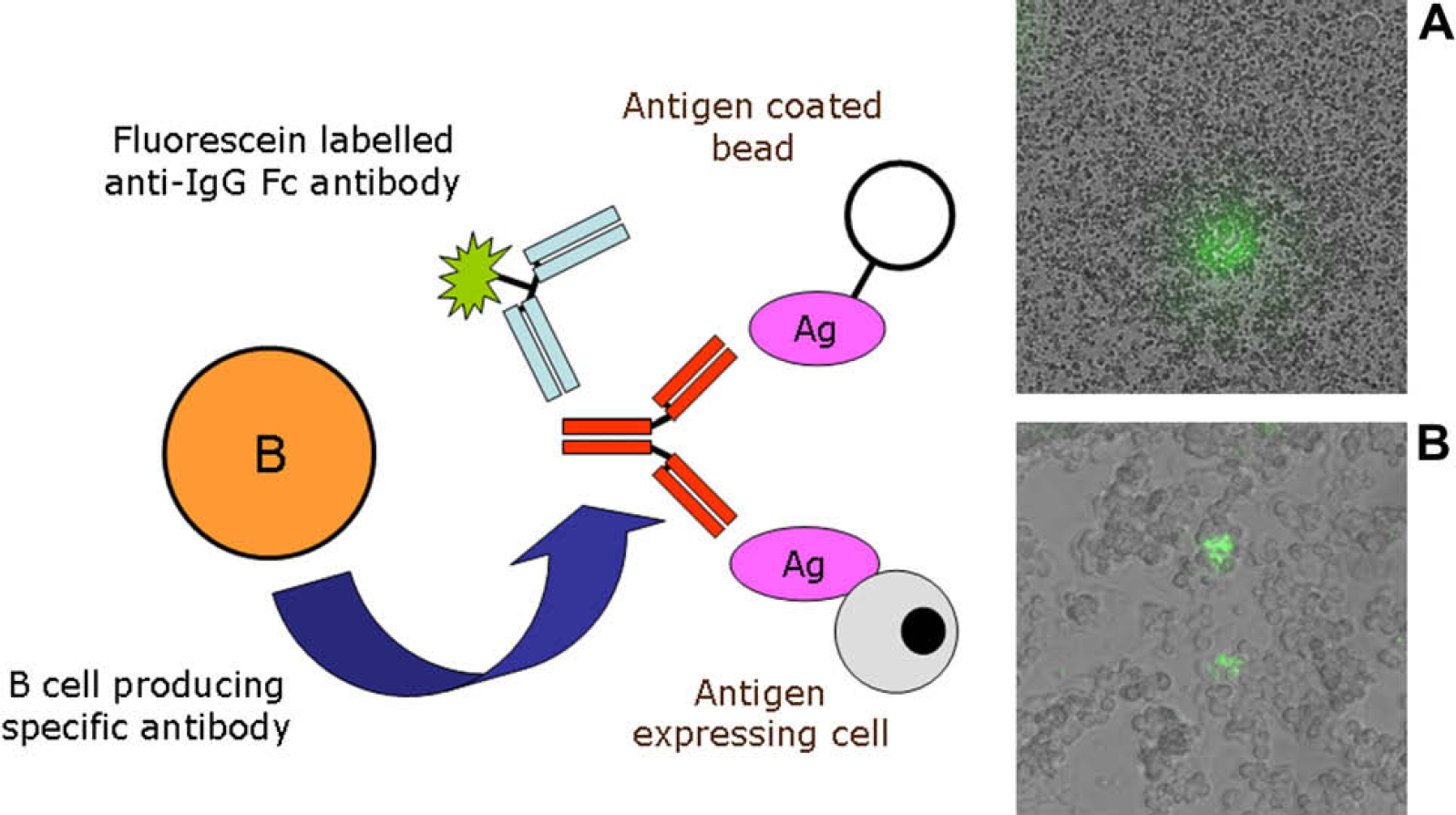
Antigen-specific B cell identification and selection. During the short culture time after thawing, the B cell secretes antibody. The specific antibody binds to either antigen (Ag) coated beads (A) or cells expressing antigen (B) in the vicinity of the B cell, producing a concentrated area of antibody. The fluorescein-labeled anti-IgG Fc antibody binds to the secreted antibody producing a halo of fluorescence surrounding the specific B cell. The images A and B show superimposed bright field and fluorescence microscope images.
Molecular Biology
From each cell picked, cDNA is prepared directly from the lyzed cells. The gene cloning process is carried out using a dedicated Aviso Onyx liquid-handling robot. Nested PCR using species specific variable region primer sets, is used to amplify the antibody heavy and light chain variable region genes 3 through two rounds of PCR. The liquid-handling robot also tracks samples using plate barcodes. Material from the second PCR reaction is used to clone the genes into transient expression vectors using standard techniques and sequence the antibody heavy and light chain variable region genes.
Transient Expression
An automated transient expression system using the Freestyle 293 Expression System (Invitrogen, Carlsbad, CA) 4 has been developed. The existing liquid-handling robots are used to speed up the setup and the harvest of material and track samples. Heavy and light chain DNA is mixed together and then 293fectin is added. The 293fectin/DNA mixture is then added to the Freestyle 293F cells. The transfected cells are cultured for between 3 and 5 days before harvesting of the supernatant. The supernatants from the transient expression cultures are used to verify the results from the primary and secondary screens.
Summary
The UCB SLAM process described here has had a positive and dramatic effect on the way we select therapeutic candidate antibodies. An example of the number of cells that can be screened using UCB SLAM is shown in Table 2. UCB previously used the hybridoma method to generate antibodies for both research and for therapy. 5,6 This method, we believed, was not particularly amenable to automation, due to the large amount of cell culture involved in both setting up the fusion and then maintaining the hybridoma cell lines, although this approach has been adopted by others. 7 The in vitro culture of B cells, combined with the process automation has allowed many more antibodies to be screened, with hybridoma only those B cells that have fused with the myeloma are available for screening, this is only a small proportion of the total number of B cells. UCB SLAM B cell cultures can be screened a week after they are set up compared with 2–3 weeks for hybridoma. Advances in the molecular biology have allowed us to move away from the time-consuming dilution cloning of cells to select antibodies; the antibody genes can be isolated, sequenced, and recombinant antibody transiently expressed in 2 weeks. The method does not require a species specific, myeloma cell line as a fusion partner, which means there is no constraint on B cell source species. The lack of a B cell fusion event means that a highly efficient sampling of the B cell repertoire can be achieved. We have found the process to be applicable to generating research reagents, high quality antibodies to animal target antigens. 8 These research reagents can be raised to different target antigen epitopes than used in both in vitro and in vivo mechanistic and disease models. The research reagents are a significant aid to investigate the complex biology of our targets. The lack of species-specific fusion partners makes this approach a challenge for hybridoma, particularly when there is a need to generate antibodies to mouse and rat antigens.
An example of the numbers involved in the Selected Lymphocyte Antibody Method (SLAM) antibody screening process

The target antigen was a human cytokine. The screening and cloning process took place over a 3-month period.
Phage display can be a rapid way of selecting antibodies. 9 The initial panning for specific antibodies may be quick but additional affinity maturation steps are then required to produce a good quality antibody. Unlike phage display, the antibodies produced using UCB SLAM come from a non-naive source; the immunized animal's immune system has already done the affinity maturation step and the antibody heavy and light chain pairings are maintained.
There is very little published work on high-throughput screening in antibody generation, whether by phage display, ribosome display, or hybridoma. 10 This makes the comparison of the different techniques to generate antibodies very difficult.
UCB SLAM is continually being refined and updated, it is hoped that these changes to the biology, molecular biology, hardware, and software will lead to a marked improvement in the antibody generation process.
Footnotes
Acknowledgment
The authors would like to thank David Lee, Matt Page, and James Snowden of UCB's Informatics Department for their expert help with the LIMS and data handling.