
Abstract
At SomaLogic, we have embarked on an ambitious program of clinical studies using a novel aptamerbased proteomics technology to discover biomarkers and develop new tools to diagnose, understand, and treat human disease. As part of this program, we designed and implemented an automated assay for its highly multiplexed proteomics discovery platform. The performance of the automated assay was validated in a study that compared the automated assay to the specifications of an established manual method. Results showed that the automated method performed to the required specifications, and that the automation system improved the efficiency, productivity, and economics of our biomarker discovery program.

Introduction
Molecular in vitro diagnostics holds the promise to transform medicine through detecting and diagnosing disease, guiding treatment, and aiding in the development of new treatments. The circulating proteome is an important hunting ground for biomarkers because protein concentrations in blood are more closely tied to disease phenotype than any other kinds of biomarkers in blood. 1 –4 However, whole-proteome proteomics has not been achieved because the ability to measure thousands of proteins, with concentrations that range over 10 logs, requires measurements with extreme sensitivity and specificity. We have thought hard about these requirements, and conclude that they are unmet currently by mass spectrometry and antibody-based methods, the two leading proteomics technologies. See more on this below, and for a thorough discussion of this critical issue, see our recent paper on the topic. 4
Our proteomics discovery assay (the assay) uses SLaptamers (SL = slow), a new class of aptamer-based binding reagent developed specifically for this technology. 5,6 Aptamers are single-stranded nucleic acid molecules that are selected to bind tightly to a specific target molecule using the in vitro selection method Systematic Evolution of Ligands by Exponential Enrichment (SELEX). 7,8 Aptamers fold into unique three-dimensional shapes that are highly stable and resistant to degradation, for example, by nucleases. SLaptamers are next-generation aptamers that are selected with novel SELEX methods and modified nucleic acids that we developed to produce a new class of protein binding reagents with exceptionally high sensitivity and specificity. With these reagents, we can multiplex up to thousands of protein measurements in a single proteomics assay for biomarker discovery. The assay measures proteins in complex matrices such as plasma and serum with exceptional sensitivity, as low as 10 fM, over a large dynamic range of at least 5 logs. 5
Consequently, over the past decade we have developed a powerful new proteomics technology, capable of large-scale proteomics, that measures hundreds to thousands of proteins in a single assay that is fast, economical, and highly scalable for biomarker discovery. Once biomarkers are discovered, we use similar technology to develop commercial in vitro diagnostic tests for clinical use. This technology is poised to do for protein biomarker discovery what DNA chip technology has done for the discovery of nucleic acid biomarkers, which includes discoveries in single nucleotide polymorphism identification and copy number variation measurements.
This level of performance has not been achieved by MS or antibody technologies. MS lacks the sensitivity to measure low abundance proteins, and is slow and expensive. 9 Antibodies lack the specificity required for multiplexed whole-proteome proteomics. 4 Furthermore, antibodies are only available for a fraction of the >3500 proteins estimated to comprise the circulating proteome. 10 –13
In contrast, we can select SLaptamer binding reagents for nearly any protein, including isoforms, with extremely high sensitivity and specificity. This is achieved through carefully controlled selection. 4 SLaptamers are selected with a combination of high affinity (<1 nM) for the target protein and very long off-rates (>30 min). A high affinity ensures that most SLaptamers bind the correct target protein in the first place, whereas a long off-rate ensures that only properly bound SLaptamer—protein pairs survive a stringent kinetic challenge (see below). Thus, SLaptamers are exquisite protein binding reagents that meet the requirements for a high-content proteomics discovery assay.
In our discovery assay, hundreds to thousands of unique SLaptamers are combined into a mixture that forms the basis of the assay. The complete assay is a novel molecular method that transforms the quantity of each protein to be measured into a proportional quantity of a specific cognate SLaptamer, as shown in Figure 1. 6 The end result of the assay is a complex mixture of SLaptamers that is representative of the proteins that were present in the original sample. Because SLaptamers are DNA molecules, this complex mixture is easily quantified by standard DNA detection techniques such as hybridization to a DNA microarray. The assay used for the experiments presented here included SLaptamers selected to measure 625 different human proteins. The assay is highly scalable and is ultimately capable of measuring the whole secreted proteome, estimated to be >3500 proteins. 10 –13
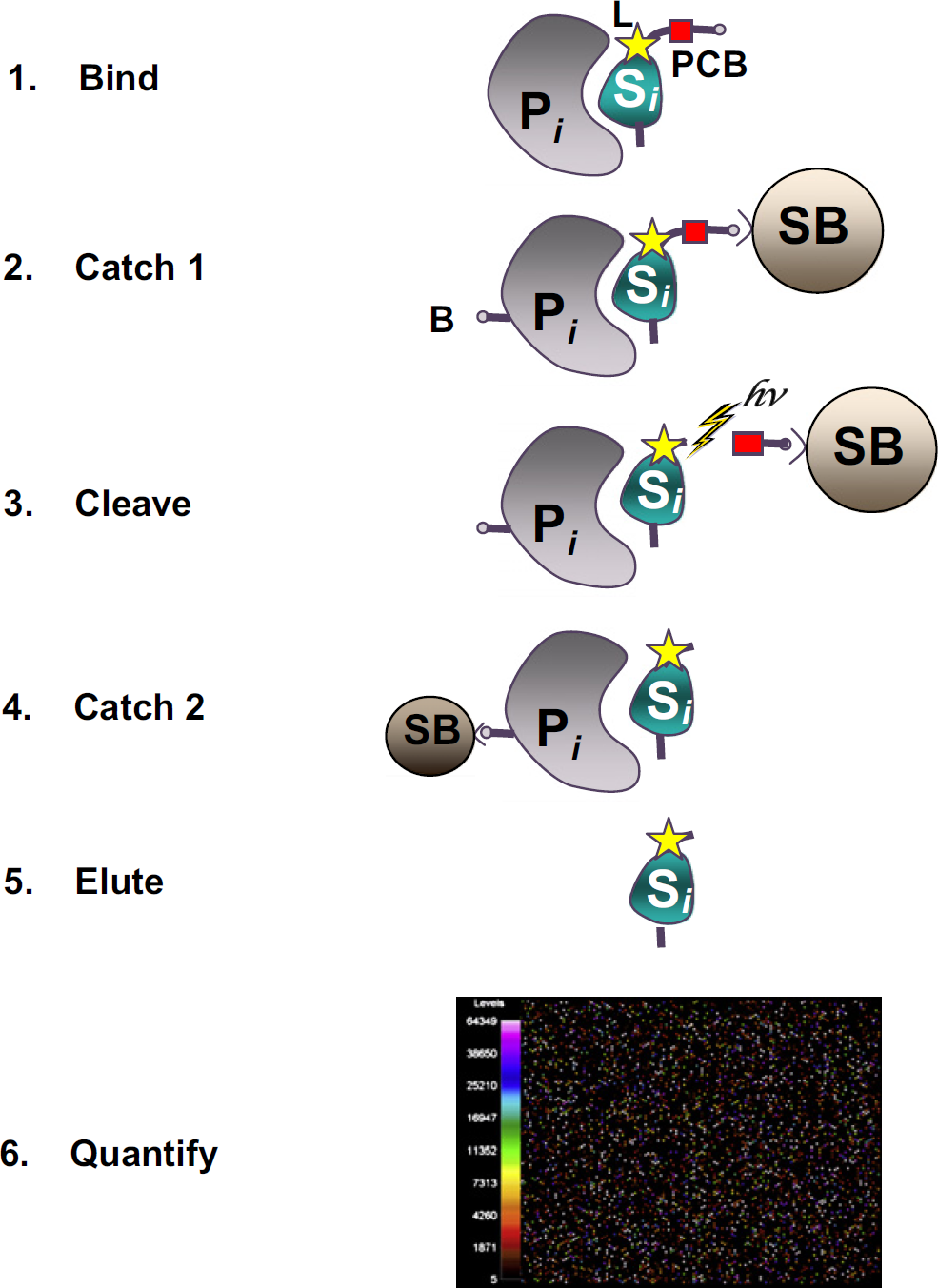
Overview of the SomaLogic proteomics assay. In step 1, the specific protein to be measured (P i ) binds tightly to its cognate SLaptamer binding molecule (S i ), which includes a photo-cleavable biotin (PCB) and fluorescent label (L) at the 5′ end. In step 2, bound protein—SLaptamer complexes are captured onto streptavidin-coated beads (SB) by the PCB on the SLaptamer. Unbound proteins are washed away. Bound proteins are tagged with biotin (B). In step 3, the PCB is cleaved by UV light (hv) and the protein—SLaptamer complexes are released into solution. In step 4, the protein—SLaptamer complexes are captured onto streptavidin-coated magnetic beads and the SLaptamers are eluted into solution and recovered for quantification in step 6, hybridization to a custom DNA microarray. Each probe spot contains DNA with sequence complementary to a specific SLaptamer, and the fluorescent intensity of each probe spot is proportional to the amount of SLaptamer recovered, and thus directly proportional to the amount of protein present in the original sample.
An important step in the development of the assay was automating the assay, which has improved its performance, efficiency, and productivity in discovering biomarkers in ongoing clinical studies of human diseases. Before automation, the prehybridization component of the assay was performed manually by a team of three people in approximately 4 h. Three important goals for the automation of the method were (1) to achieve the established performance criteria, (2) to minimize sample-handling errors, and (3) to reduce the personnel required to run the assay. Here, we present results from a validation study of the automated system and a basic economic analysis.
Materials and Methods
Automation Development
The design requirements for the automation of the assay are diagrammed schematically in Figure 2. To automate the assay, we chose a dual arm Biomek FXp robotic workstation (Beckman Coulter, Fullerton, CA), which is shown in Figure 3. We configured the Biomek workstation with two 96-channel multipipettors with grippers and an integrated ambient Cytomat microplate hotel (Beckman Coulter Part# 394597, Fullerton, CA). The Biomek's open deck format is highly flexible and easily accommodates specific automated labware positioners (ALPs) in a configuration that allows the simultaneous processing of three 96-well plates. The assay incorporates vacuum filtration, magnetic separation, and controlled orbital shaking. Shaking is accomplished with three Orbital shaker ALPs (Beckman Coulter Part# 379448, Fullerton, CA, Fig. 3).
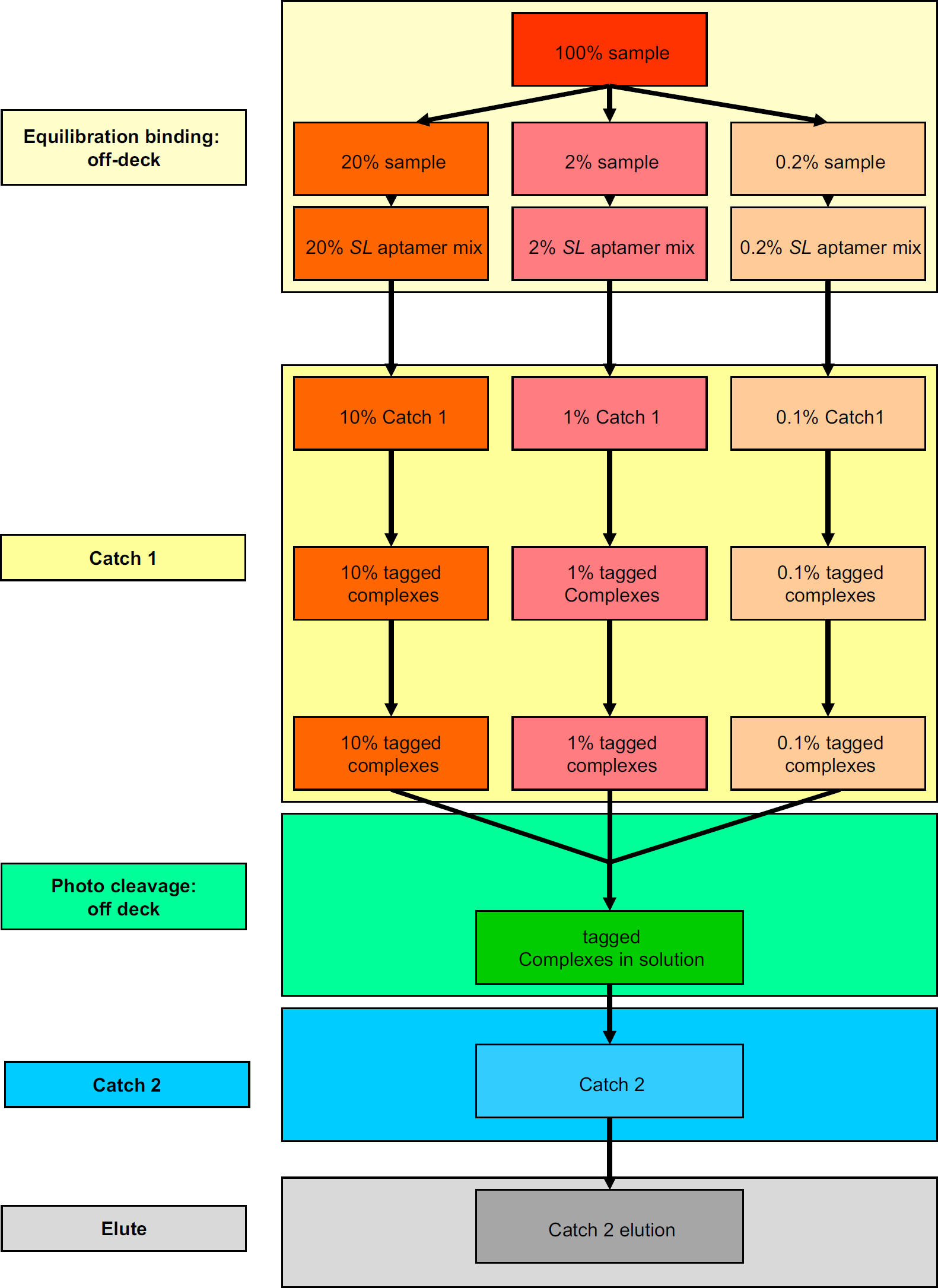
Schematic diagram of the automated SomaLogic proteomic assay. Blood samples are split into three separate dilutions and aliquoted into three separate 96-well plates. Dilution-specific SLaptamer mixtures are added. Equilibration binding is done off deck in an MJ Tetrad. This step takes 20 min. Catch-1 takes place in independent Millipore filter plates on streptavidin-coated beads shaking on the orbital shakers. Extensive washing after capture is accomplished through the three Millipore vacuum stations. Biotin tagging occurs within the filter plates incubating while shaking on the orbital shakers. Post tagging washes are accomplished by vacuum filtration. Photo cleavage is done off deck in three separate filter plates. Sample dilutions are combined into a deep-well plate by centrifugation after photo cleavage. This step takes 10 min. Catch-2. Combined dilutions are captured onto streptavidin-coated magnetic beads shaking on orbital shaker. Postcapture washes are done by magnetic separation using a donut magnet and orbital shaker. Elute. Aptamers are eluted from beads by high pH incubation on the orbital shakers. Elution is neutralized following transfer to a new plate.

Biomek FXP robot and hotel. (A) Orbital Shaker automated labware positioner (ALP), (B) Millipore HTS Vacuum Manifold, (C) Cytomat Microplate Hotel, (D) Cytomat Conveyor ALP (unload position), (E) Multimek Circulating Reagent Reservoir, and (F) Trash ALP.
Vacuum filtration is managed by three integrated Millipore HTS vacuum manifolds (Millipore Part# MSVMHTS00, Billerica, MA, Fig. 3) plumbed to three Millipore WP6111560 vacuum pumps. We chose this design for its flexibility and relative ease of control. Vacuum stations are independently controlled by the Biomek software (version 3.3.14) through a Biomek FX Device Controller (Beckman Part# 716366). The switching capacity of the Biomek FX Device Controller is below the amperes required by the vacuum pumps, so electronic relay switches (Green Air Products, IR-1, Gresham, OR, Model) controlled by the Biomek Device Controller provide power to the pumps. Orbital shaker ALPs and vacuum manifolds are positioned at strategic locations to allow seamless processing of plates without triggering delays caused by resource conflicts between the two pods.
The second purification step of the assay (Catch-2) incorporates magnetic separation. Streptavidin-coated magnetic beads are used to capture biotin-tagged complexes. The captured complexes are washed by separating the magnetic beads with a SBS format 96-well magnetic ring magnet (Ambion Part# 10050, Austin, TX) on deck. The beads are resuspended during washes on the orbital shakers.
The tips and reagents that the assay requires are loaded on the Cytomat microplate hotel at the beginning of the assay. Tips and reagents are shuttled onto the Biomek deck by the Cytomat Conveyor ALP (Beckman Coulter, Fullerton, CA) positioned so that both pods have access to the unload position. The Cytomat microplate hotel has a capacity of either 63 tip boxes, 63 reagent reservoirs, 189 lidded micro-titer plates, or any combination of the three. This capacity allows flexibility in programming methods and coordinated use of Biomek deck space when integrating many fixed equipment spaces on the deck. Because multiple steps in the assay use a common reagent, we added a Multimek Circulating Reagent Reservoir (Beckman Coulter Part# 148026, Fullerton, CA) to improve deck layout and assay speed compared with storing multiple buffer reservoirs in the Cytomat microplate hotel. A Trash ALP (Beckman Coulter Part# 719347, Fullerton, CA) is integrated into the workstation for effective waste disposal.
Method programming was accomplished using the Biomek software (Beckman Coulter v3.3.14, Fullerton, CA) installed on a Pentium 4 HT IBM ThinkCentre computer running Windows XP operating system. In house methods were developed using the software's graphical user interface and additional Microsoft Visual Basic scripting. After completion of the automated assay, samples were removed from the deck, hybridized to custom DNA microarrays, and quantified with a microarray reader.
The plasticware used in the assay included the following. Catch-1 and all filtration steps used Millipore MultiScreen HTS, HV 0.45-μM Hydrophilic Durapore filter plates (Millipore Part# MSHVN4450, Billerica, MA). Catch-2 and all magnetic steps used 0.5-mL polypropylene Nunc round-bottom plates (Thermo Fisher Scientific, Rochester, NY, # 267245). Reagents were contained in deep-well Fisher Matrix Automation Reservoirs (Thermo Fisher Scientific Part# 1064–05–6 Rochester, NY), lidded with Matrix automation-friendly microplate lids (Thermo Fisher Scientific Part# 4954 Rochester, NY). All pipette tips were Beckman Coulter tips: Biomek AP96 P250 Barrier (Beckman Coulter Part# 717253, Fullerton, CA); Biomek AP96 P250 (Beckman Coulter Part# 717252, Fullerton, CA); and Biomek AP96 P20 Barrier (Beckman Coulter Part# 717256, Fullerton, CA).
Validation Study
To validate the automated version of the assay, we compared the automated assay directly to the established manual assay in a controlled experiment that measured an identical set of serum samples. We collected 20 serum samples from healthy normal volunteers in BD SST Tiger Top Tubes (BD, Franklin Lakes, NJ). The collected blood was allowed to clot at room temperature for 30 min, and then centrifuged at 1200 × g for 10 min. The resulting serum was aliquoted into cryotubes, which were frozen and stored at −80°C until use.
For the validation study, aliquots of the 20 serum samples were assayed in triplicate with both the manual and automated versions of the assay. To measure proteins present at different abundances in serum, samples are diluted to 10%, 1%, and 0.1% (final concentrations) and mixed with three different SLaptamer mixtures. Each mixture is designed to measure proteins at that sample dilution based on their expected concentrations. The three dilutions are run separately through the first step of the assay, Catch-1, in parallel and combined before the second capture step, Catch-2. For each serum sample assayed, the resulting SLaptamer mixture is quantified by hybridization to a custom Agilent DNA micro-array (Agilent, Santa Clara, CA) that contains 10 replicate probe spots of complementary DNA for a unique portion of each SLaptamer, as well as probe spots complementary to internal hybridization controls. SLaptamer mixtures were hybridized for 17 h, washed, and read with a microarray reader (Agilent Model G2505C, Santa Clara, CA). 6 The resulting raw scanner data was processed and analyzed as described in the following section.
Hybridization Normalization
The quantity of SLaptamer hybridized to each cognate probe spot on the microarray is measured by the scanner in raw fluorescent units (RFUs). Changes in RFU signal strength can occur between samples due to factors such as inconsistent hybridization conditions and laser power fluctuation in the scanner. To compensate for such changes, we normalize RFU values for all SLaptamers on an array using internal hybridization controls added just before hybridization.
Assay Normalization
Assay normalization is performed to reduce signal variation potentially introduced during the assay. Each sample in a study is normalized using a set of normalization SLaptamers that have the lowest overall relative signal variation across all samples within a study. For each normalization SLaptamer, its median value is calculated from all samples in the study, and together these median values are used to calculate a scaling factor for each individual sample. The scaling factor is the mean of a series of values, one for each normalization SLaptamer, calculated as the sample signal divided by the median signal for the study. When applied to a sample, this procedure brings the signals corresponding to SLaptamers in the normalization set closer to the median values across the assay, and reduces the observed variation between replicate samples for all SLaptamers.
Dilution Normalization
Because the assay splits each serum sample into three dilutions, assay normalization was performed separately on the three SLaptamer groups corresponding to the 10%, 1%, and 0.1% dilutions. Dilution normalization applies the same constant factor to every signal from any given sample. This factor varied between samples in the range from 0.8 to 1.2, and is typically within 10% of unity.
CV Calculations
To compare the variability of the manual and automated assay, we calculated coefficients of variation (CVs) for each SLaptamer from the three replicate samples for each of the 20 individuals in the study. The overall CV of a single SLaptamer signal for an individual was calculated as the ratio between the sample standard deviation and the mean of the measurements from the nine repeated samples from the serum of a given individual. This includes variation between the three repeats within each assay run and variation between the three independent runs of the assay. The interrun CV values reported here for each SLaptamer were calculated as the mean of the 20 individual estimates of overall CV of the SLaptamer signal.
Results and Discussion
The assay is a multistep process that involves two capture steps (Catch-1 and Catch-2), as diagramed in Figure 1. Before automation of the assay, it was performed manually in a 96-well format by a team of three people in 4 h. Teams ensured proper timing for the simultaneous pipetting of the three 96-well plates used in Catch-1 (see Fig. 2). The automated version of the assay takes 3 h to complete with much less manual labor. The automation of the assay was accomplished with off-the-shelf hardware and software. The design of the automation system was based on the following criteria:
Accommodate three 96-well plates simultaneously with precise timing;
Three vacuum filtration stations;
Three orbital shakers;
Magnetic separation; and
Large hotel for external storage of materials.
Based on the design criteria, we chose the Biomek FXp robotic workstation over other systems surveyed. Cost was a factor but flexibility and sample processing capabilities were also important considerations. The first design criterion, the ability to process three plates simultaneously with precise timing, led us to choose the dual arm multichannel Biomek FXp workstation. The Biomek's open deck format allowed for highly flexible incorporation of additional devices. The Biomek software was user friendly, with most method programming accomplished through a graphical user interface, but also flexible enough to allow for custom scripting to accomplish some nonstandard operations. Beckman software is uniform across all of their liquid handlers, which allows knowledge and resources accumulated to be applied to future Beckman systems. Finally, the Biomek series is a well-established platform across the biotechnology industry.
Standard performance testing showed that the assembled system performed to its technical specifications for functions such as precision and accuracy of liquid delivery. The development of the multistep method for the assay was also achieved with relative ease using the Biomek software's graphical user interface, which allowed for easy control of the Biomek workstation and all associated ALPs. A few of the more complicated method parameters, such as controlling vacuum filtration stations, were accomplished with custom Visual Basic scripts. Whole system pauses, as well as command prompts, were used to manage two off-deck steps in the assay, as shown in Figure 2.
To test the performance of the automated assay, results were compared from a validation study run in parallel using the manual and automated assay methods. The validation study included serum samples collected from 20 healthy individuals. For each run of the assay, serum samples were assayed in triplicate for each of the 20 individuals. For the study, the assay was run three times each for the manual and the automated method. Thus for each individual, the levels of 625 proteins were measured nine separate times for both the automated and manual methods.
The result of each serum sample run through the assay was a solution containing a mixture of SLaptamers, each at a concentration proportional to its cognate protein in the original serum sample. To quantify the results, each mixture was hybridized to a custom Agilent DNA microarray and read on an Agilent microarray reader. The resulting raw data, expressed in relative fluorescent units (RFUs), was normalized and analyzed to determine the variation in the assay. These results were used to compare the precision of the assay performed with the manual and automated methods.
To compare the precision of the manual and automated assays, intra- and interrun average CVs were calculated for each of the 625 protein measurements made for each sample in the validation study. Figure 4 shows the distributions of the intrarun CVs for the automated and manual assays. The median intrarun CVs for the two methods are the x-values that correspond to the middle of the y-value range (approximately 100). The median intrarun and interrun CVs for the two methods are shown in Table 1. These results show that the precision of the automated assay met or exceeded that of the manual assay, and therefore, the precision performance goals set for the project were achieved.
Precision of proteomics assay

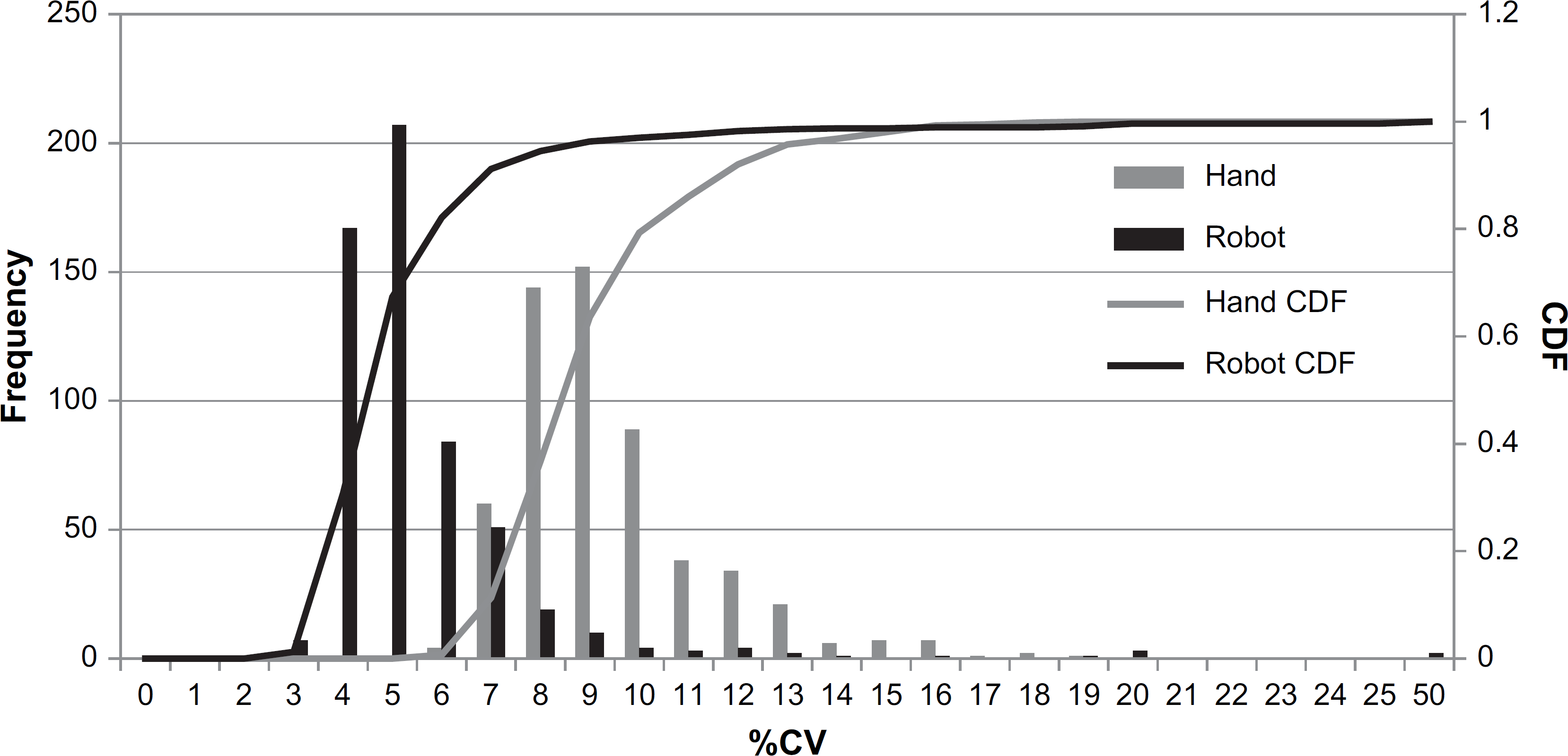
The distribution of %CV for the automated assay (black) and the manual assay (gray) shown as histograms and cumulative distributions (CDF). Values shown are the intrarun CVs calculated from raw fluorescent unit data that was normalized with the hybridization normalization method.
Because serum protein concentrations vary from person to person, the CVs cannot be compared across all samples as a measure of variability in the assay. Instead, CVs were calculated for the three repeated samples for each individual in an assay run, and these CVs were averaged between the 20 individuals to provide estimates of the intrarun CV for each SLaptamer. For each SLaptamer, the intrarun CV was calculated as the mean of the 60 estimates (20 individual CV estimates from each of the three independent assay runs). The overall CV reported was calculated as the mean for all 625 SLaptamers across the study.
To compare the precision of the automated and manual assays, two normalization methods were applied to the data. In the first method, hybridization normalization, the total RFU signal strength for each sample was normalized across the entire study (detailed in Materials and Methods). Such normalization reduces the coefficients of variation across repeated sample measurements. Because the sources of the hybridization and scanning variation that are addressed by hybridization normalization are due to factors downstream of the assay methods (manual and automated) described in this paper, hybridization normalization was performed on all the measurements reported here. Normalization of this form applies the same constant factor to every signal from any given sample. The hybridization normalization factor varied between samples in the range from 0.8 to 1.2, and was typically within 10% of unity.
The second normalization method used, assay normalization, was performed to further reduce signal variation potentially introduced during the assay, such as variation due to small volume differences in pipetting samples, for example. Because assay normalization is intended to reduce the variability of the measurements caused by steps in the assay, the results from both with and without such normalization are reported.
To directly compare results from the automated and manual methods, the average RFU value for each SLaptamer automated run was correlated to the corresponding value from the manual run, as shown in Figure 5. There was a strong correlation between the automated and manual assays (R 2 = 0.966), which shows that there was no statistically significant difference between the automated and manual assays. Similarly, a run-to-run correlation for the automated assay showed no significant difference (R 2 = 0.976), as shown in Figure 6.
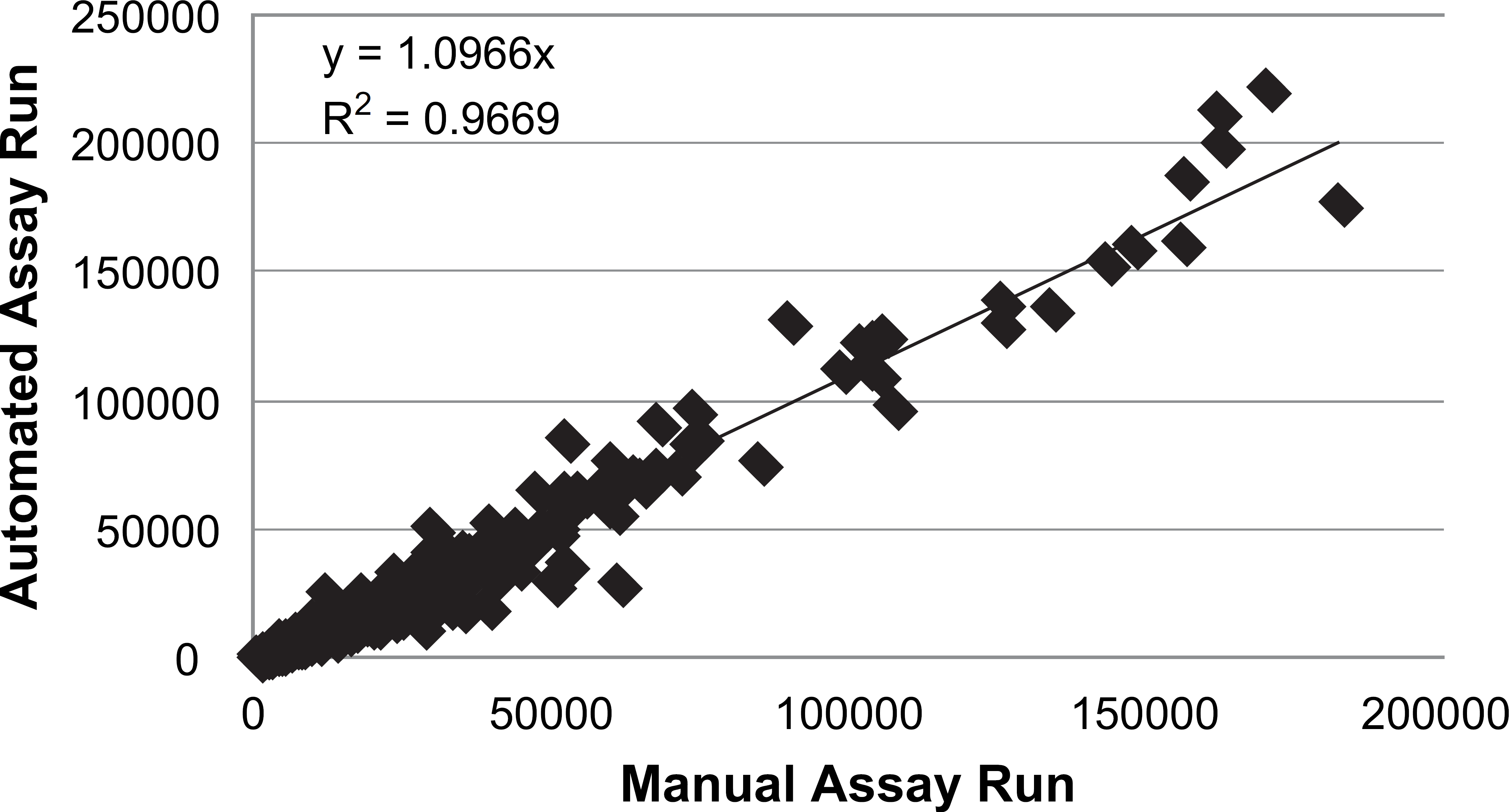
Correlation of automated and manual assay, comparing average raw fluorescent unit values for 625 SLaptamers. Each value is the average from triplicate runs of 20 individuals.
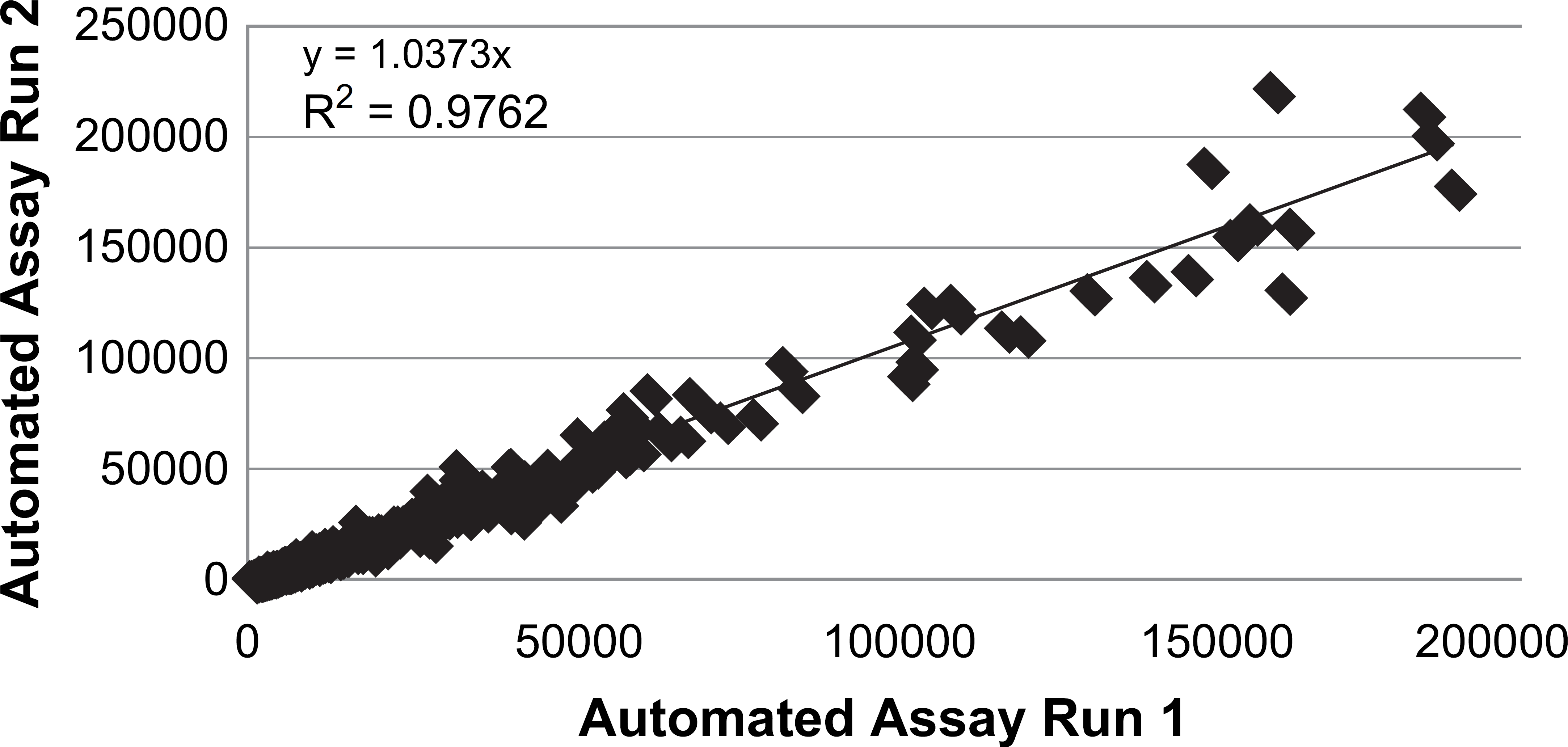
Correlation of duplicate automated runs, comparing average raw fluorescent unit values for 625 SLaptamers calculated from 20 individuals.
The automated assay was validated further with clinical sample sets and is now running in full production mode in our laboratory. In a recent clinical study of patients with lung cancer, we used the automated assay to measure 625 proteins in nearly 1000 clinical samples, all in duplicate, in 2 weeks. Thus, we ran the equivalent of >106 ELISAs (Enzyme-Linked Immunosorbent Assays) in that short time. With the automated system's current configuration, the assay is completed in 3 h and we have a capacity of approximately 50,000 samples per year. This capacity can be expanded easily by adding additional robotic workstations. An analysis of the impact of the automated system on the workflow and human resources of the assay team showed that fewer personnel accomplished the same throughput with an easier workflow and less human error than the manual version of the assay.
In conclusion, we have developed an automated system and assay for its proteomics discovery platform. The automated assay was validated in our production laboratory and has performed consistently without error. Finally, a cost—benefit analysis showed a substantial benefit from the automated system in terms of real cost savings and increased production. Taken together, the results show that the automated assay met the performance criteria established for its development. Our automated proteomics platform is now in full operation and is improving the efficiency and productivity of our ongoing efforts to help transform medicine through the creation of evidence-based tools that detect and diagnose disease, guide treatment, and aid in the development of new therapies.
Acknowledgments
We thank everyone at SomaLogic for their contributions, which ultimately have made this work possible. We especially thank Trudi Foreman and Meredith Goss for the collection and handling of serum samples for the validation study, and Dom Zichi, Britta Singer, Sally Nelson, Diana Maul, and Kirk Harris for their critical review of this manuscript. This research was paid for by SomaLogic, Inc. and all authors are employees of SomaLogic, Inc.