
Abstract
Emerging molecular studies have shown that the transcription factor NF-E2-related factor (Nrf2) plays an essential role in cancer chemoprevention. Here, we report the development of a molecular biosensor for rapid detection of antioxidant-responsive element (ARE)-bound Nrf2 protein. The development will provide a molecular assay for high-throughput screening of chemopreventive compounds. Specifically, a double-stranded DNA probe is designed based on the ARE sequence. One of the DNA strands is labeled with a fluorophore on the 5′ end and the complementary strand is labeled with a quencher on the 3′ end. A single-stranded DNA competitor is also designed. The existence of the Nrf2 stabilizes the fluorescent probes and delays the competitor from separating the fluorophore-quencher complex. Therefore, the concentration of the Nrf2 proteins can be measured quantitatively based on the fluorescence intensity. The molecular binding scheme was demonstrated using purified p50 and the detection of endogenous Nrf2 was demonstrated using whole-cell lysates treated with sulforaphane. The assay can easily be incorporated into an automated platform for high-throughput screening of chemopreventive compounds targeting Nrf2.
Introduction
Various environmental insults, such as toxicants and carcinogens, translate into oxidative stress or electrophiles at the molecular level. In dealing with the toxicant- and carcinogen-induced oxidative stress or electrophiles, cells have evolved defense mechanisms by inducing the expression of phase II enzymes and endogenous antioxidants to defend cells from oxidative stress and reactive carcinogenic intermediates. Some of the well-known cytoprotective enzymes include NADPH:quinone oxidoreductase 1, heme oxygenase-1, and γ-glutamylcysteine synthetase. 1 In general, emerging evidence has shown that the expressions of many cytoprotective enzymes are regulated by NF-E2-related factor 2 (Nrf2) via antioxidant-responsive elements (AREs) and many chemopreventive compounds exert their effects by inducing Nrf2 activity. 1 -5 Therefore, targeting Nrf2-mediated gene expression represents one of the prominent strategies for cancer chemoprevention. Interestingly, the discovery of Nrf2 is attributed mostly to studies with anticarcinogenic compounds.
The core of the Nrf2 dynamic regulation is the Keap1-mediated degradation of Nrf2. Figure 1 presents the dynamic regulation of Nrf2. At basal conditions, Keap1 actively targets Nrf2 for ubiquitination and proteasomal degradation, resulting in short half-life of Nrf2. 4,5 On exposure of cells to Nrf2 inducers, such as chemopreventive agents, the Nrf2-Keap-1 complex disassociates and the half-life of Nrf2 increases significantly. On the other hand, Nrf2 mRNA levels were unaffected by various Nrf2 inducers. 4 -6 The level of Nrf2 is, therefore, controlled primarily though a dynamic balance of Nrf2 synthesis and degradation. As Nrf2 regulates the antioxidant response via ARE-mediated downstream target genes, the level of the ARE-bound Nrf2 protein can provide a direct indication of the cellular antioxidant response. The ability to rapidly detect the ARE-bound Nrf2 protein, therefore, represents a key step toward high-throughput screening of chemopreventive agents. 7
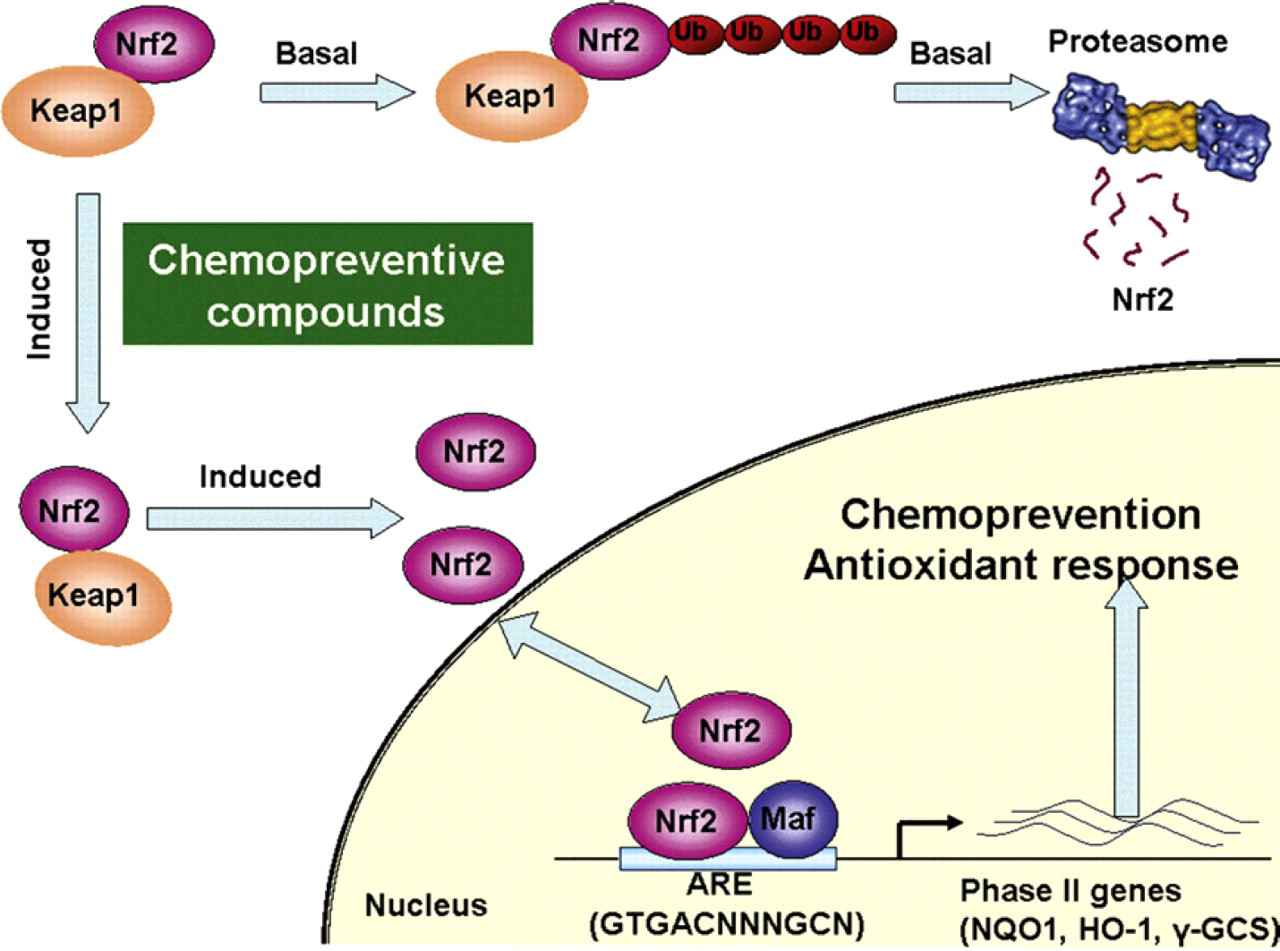
Schematic of the Nrf2-Keap1 dynamic regulation.
Electrophoretic mobility shift assay (EMSA) and reporters gene assays are currently applied for measuring the Nrf2 protein. 8,9 These assays, however, could be time/labor/cost intensive for high-throughput and multiplex applications. These shortcomings present major roadblocks for performing a high-throughput screening of compounds targeting the Nrf2 protein level. A simple, convenient, and high-throughput detection scheme for transcription factors is therefore highly desirable. 7 Several homogeneous assays have been developed for studying protein–DNA interactions. 10 -14 Nevertheless, these assays have not been optimized for detection of the ARE-bound Nrf2 protein and high-throughput screening of chemopreventive compounds. Here, we report the development of a homogeneous assay (i.e., mix and measure) for Nrf2 that are well-suited for high-throughput screening as they avoid cumbersome and time-consuming steps, such as washing, filtration, and separation.
Design Concept
The molecular biosensor takes advantage of the specific interaction between a DNA-binding protein and its corresponding binding sequence. In this molecular design, a double-stranded DNA probe is designed based on the binding sequence. The probe is labeled with a fluorophore and a quencher (Fig. 2). A complementary single-stranded DNA competitor is also synthesized. In the absence of the target DNA-binding protein, the competitor separates the fluorophore and the quencher and fluorescence intensity can be detected. In the presence of the protein, the molecular binding stabilizes the double-stranded DNA probe. The amount of the stabilized fluorescent probes should correlate to the amount of DNA-binding proteins. Therefore, the concentration of the target molecular can be measured quantitatively based on the fluorescent intensity. The molecular binding scheme provides an effective approach for rapid detection of Nrf2 and other DNA-binding proteins.
Materials and Methods
We designed two set of probes to evaluate the molecular binding scheme. Table 1 summarizes the probe sequences of the molecular biosensor in this study. A double-stranded DNA probe was designed according to the NF-κB binding sequence (GGGACTTTCC). Two bases were added to avoid interference between the NF-κB protein and the fluorophore-quencher pair. The equilibrium-binding affinity of the probe can be adjusted by modification of the remaining sequence. Similarly, a molecular probe for detecting Nrf2 proteins was designed using the ARE sequence (GTGACTCAGC). Fluorescein (6-FAM) and Iowa Black FQ were selected as the fluorophore-quencher pair. DNA probes were synthesized by Integrated DNA Technologies Inc. Purified p50 proteins were purchased from Active Motif Inc. Other reagents were purchased from Sigma. DNA probes were prepared in a buffer solution consisting 10 mM Tris-HCl and 100 nM NaCl. Before the experiment, the probes were incubated at 90 °C for 5 min and slowly cooled to room temperature in a dry bath incubator. Quantitative fluorescence measurements were taken in 384-well plates (Corning Inc) using a microplate reader (BioTek, Synergy 2).
Molecular probe designs for NF-κB and Nrf2 sensing

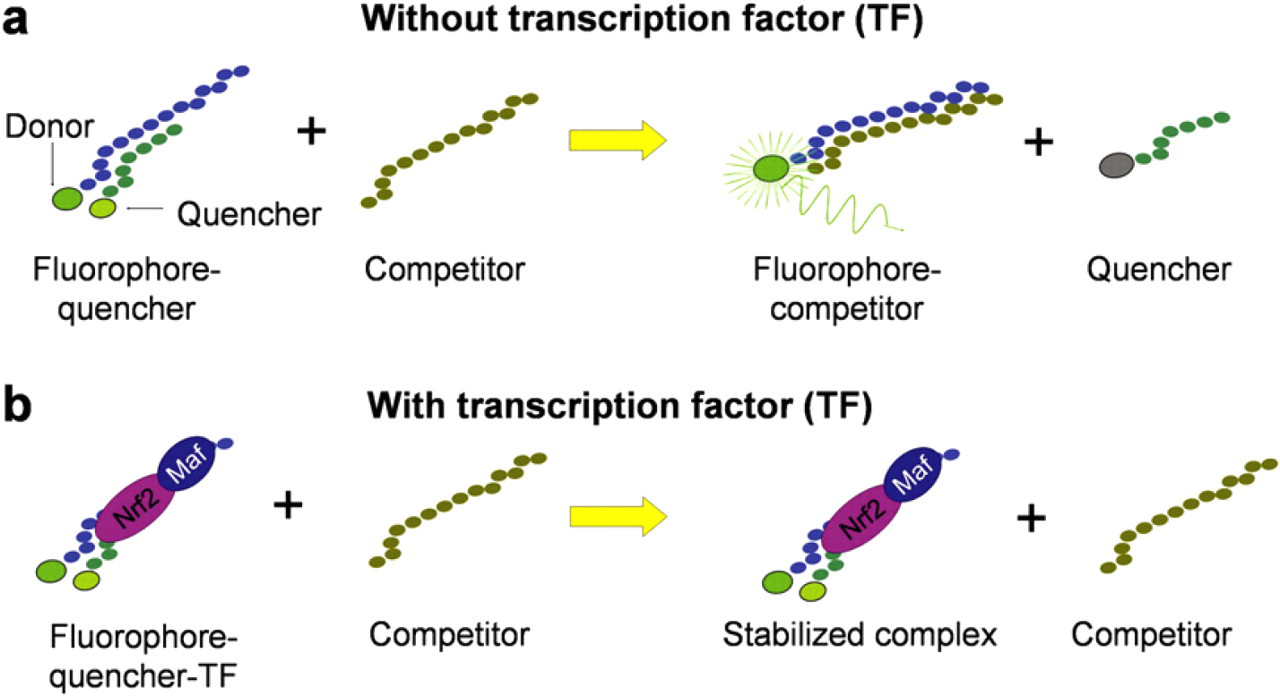
Schematic of the sensing scheme for homogeneous detection of DNA-binding proteins.
For the NF-κB experiment, the binding buffer consisted of 10 mM Tris-HCl, 50 mM NaCl, 3 mM MgCl2, 10% glycerol, 0.5 mM dithiothreitol (DTT), and 0.5 mg/mL bovine serum albumin (BSA). For the Nrf2 experiment, MDA-MB-231 cells were used. Monolayers of MDA-MB-231 cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The cells were incubated at 37 °C in a humidified incubator containing 5% CO2. The experimental procedure was designed based on our previously reported protocol. 15 Briefly, the cells were treated with 7.5 μM sulforaphane (SF) before the experiment. The cells were then washed twice with phosphate buffered saline (PBS) and carefully scraped off. The cells were lysed in a lysis buffer containing 20 nM Tris-HCl, 150 mM NaCl, 1 mM ethylene diamine tetraacetic acid (EDTA), 1 mM ethylene glycol tetraacetic acid (EGTA), 0.2% NP-40, 1 mM DTT, 1 mM phenylmethanesulphonylfluoride (PMSF), 1 mM Na3VO4, 10% glycerol, and proteinase inhibitors. The cell lysates were then centrifuged at 14,000 rpm for 10 min. The supernatants were collected and diluted with a binding buffer containing 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 60 mM KCl, 2 mM MgCl2, 1 mM EDTA, 0.004% NP-40, 10% glycerol (vol/vol), 1 mM DTT, and 2 μL Tris EDTA.
Results
Probe Optimization
In the molecular assay, the concentration of the quencher probe strand relative to the concentration of the fluorophore probe strand can be flexibly adjusted to maximize the signal-to-noise ratio. This represents an advantage of the molecular assay over other related molecular probes for DNA-binding proteins. We performed an experiment to determine the optimal quencher-to-fluorophore ratio. In the experiment, the fluorophore probe stand was maintained at 20 nM and different concentrations of the quencher probe strand were added to obtain different quencher-to-fluorophore ratios. The probes were incubated at 90 °C for 5 min and slowly cooled to room temperature for 2 h. Figure 3 shows the results of the experiment. The resulting data indicated that a 3:1 ratio was able to obtain a low noise level for the probe (Fig. 3). Further increase in the concentration of the quencher strand did not reduce the background intensity. It is consistent with other studies with similar probe designs. 16,17 Therefore, all the subsequent experiments were performed using this fluorophore-to-quencher ratio.
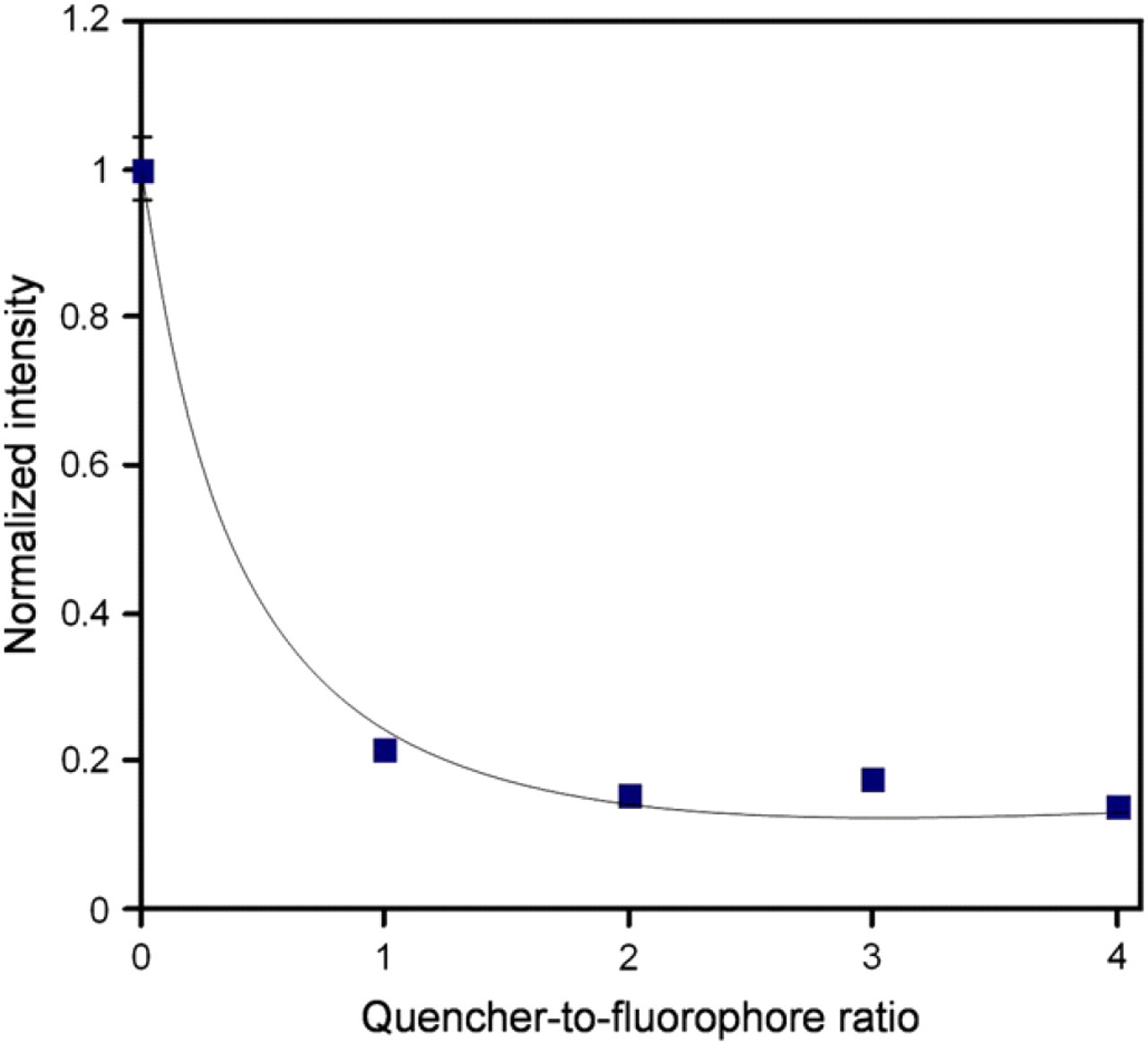
Optimization of the quencher-to-fluorophore ratio. The concentration of the fluorophore probe strand was 20 nM and the concentration of the quencher probe strand was adjusted to obtain different quencher-to-fluorophore ratio. Error bars, which are smaller than the symbols, represent the standard deviation.
NF-κB Detection
To evaluate and optimize the performance of the molecular assay, experiments were performed using purified p50 proteins (NF-κB). We chose NF-κB based on its relatively well-known structure, regulation, function, and its importance in inflammation. 18 -22 In the experiment, the probe was incubated with the protein for approximately 1 h. The competitor probe was then introduced into the well. The fluorescence intensities were monitored as a function of time after the addition of the protein and the competitor for over 2 h. Figure 4 shows the result of a representative experiment. After the protein was mixed with the probe, a small decrease in fluorescent intensity was observed. It is likely that the p50 protein stabilizes the probe and reduces the fluorescent intensity. Upon the addition of the competitor DNA, the fluorescent intensity increased rapidly, which indicated separation of the fluorophore and quencher pair. Figure 5 shows the effect of the concentration of p50 on the fluorescence intensity at different times. We observed a concentration dependence of the p50 protein on the fluorescence response. The limit of detection is on the nano molar range. The fluorescence intensity decreases with increasing concentration of p50. Therefore, the concentration of p50 can be determined based on the fluorescence intensity.
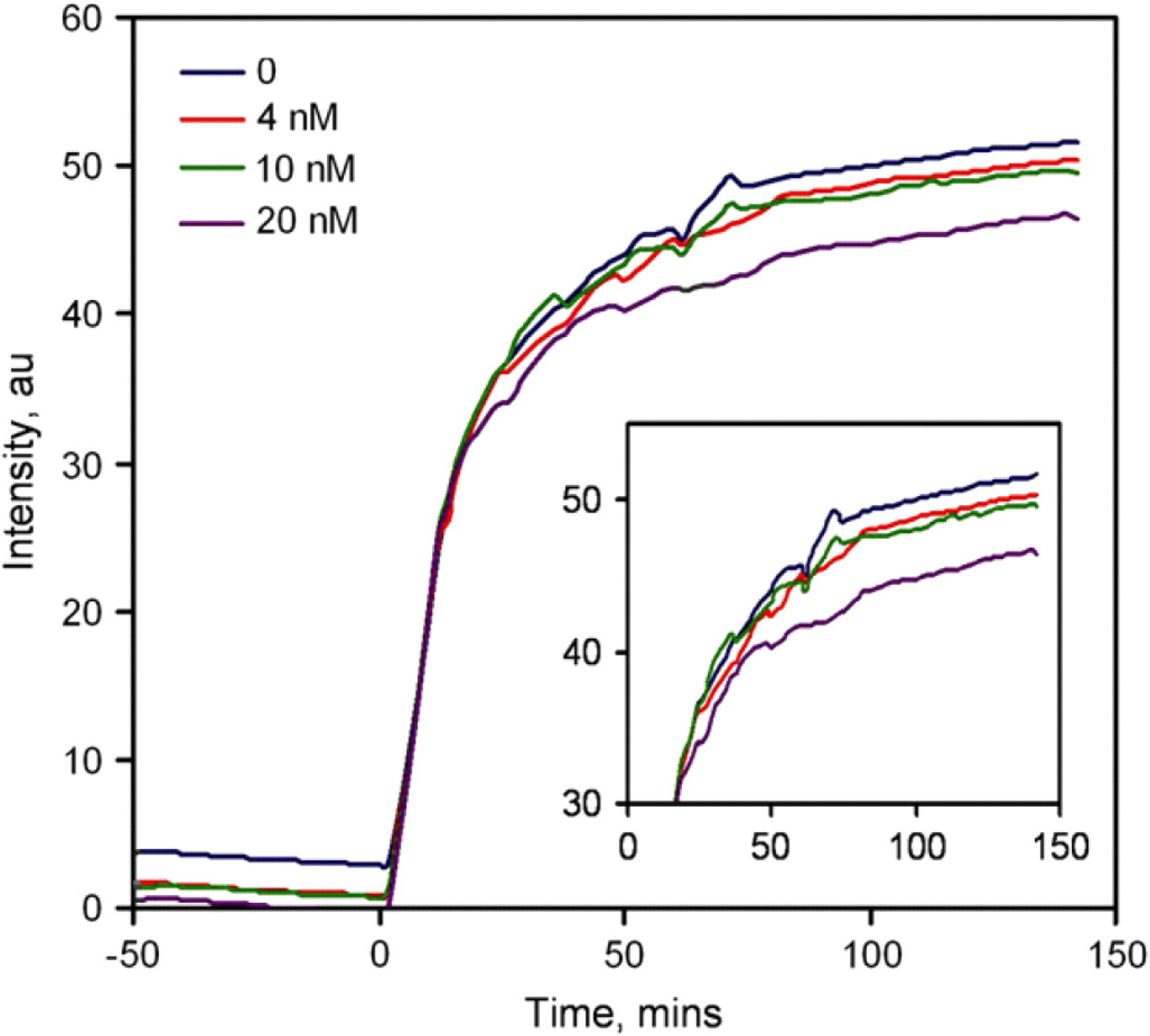
Kinetics of the NF-κB-binding assay. Different concentrations of p50 proteins were incubated with the probes for 1 h. DNA competitors were added at time zero. The sample consisted of 20 nM of fluorophore probe, 60 nM quencher probe, and 20 nM competitor. (Insert) A close-up view of the data.
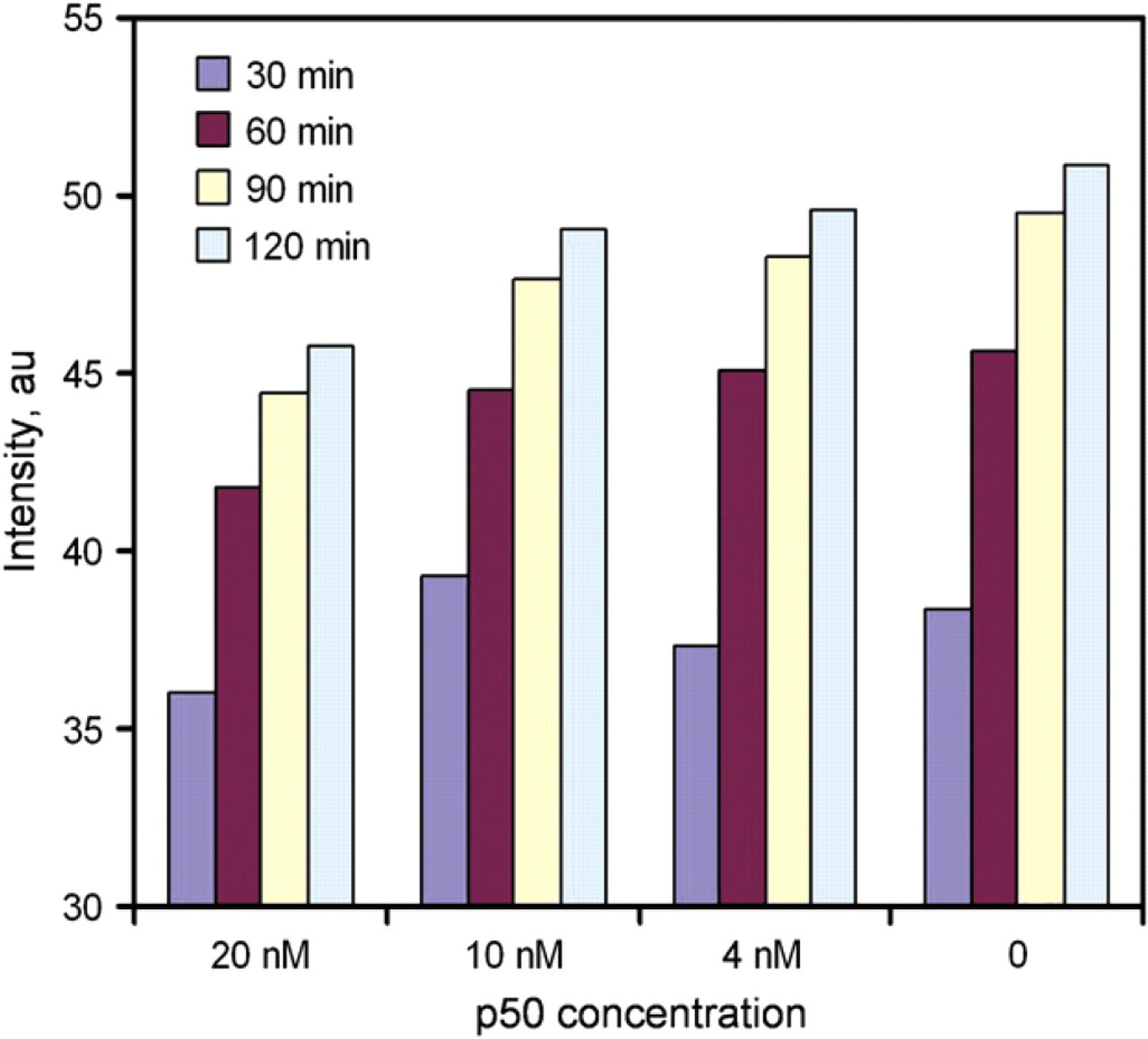
Rapid quantification of NF-κB using the molecular probe.
Nrf2 Detection in Cell Lysates
The goal of this work was to develop a rapid molecular assay for detecting ARE-bound Nrf2 proteins. The molecular biosensor will form the technological core of a cell-based assay for high-throughput screening of chemopreventive compounds. Therefore, we tested the molecular assay using whole-cell lysates. MDA-MB-231 human breast cancer cells were chosen as the model system. The cells were treated with SF, a well-known anticancer compound. 4,5 It is known that SF upregulates the activity of Nrf2, which provides an ideal model to evaluate the molecular assay for rapid screening of chemopreventive compounds. Cells treated with buffer only were used as control. Figure 6 shows the result of the experiment. We observed a rapid increase in the fluorescence intensity for the control cell sample, similar to the NF-κB experiment. SF-treated cells, on the other hand, showed a slower increase in fluorescence intensity and the control sample had a higher intensity compared to SF-treated cells at all time points. It is consistent with our hypothesis that the protein–DNA interaction stabilizes the probe and delays the increase in fluorescence intensity. At low concentration of the Nrf2 protein, the competitor rapidly separates the ARE probe and raises the fluorescent intensity. In the present of the Nrf2 protein (cells treated with SF), the molecular interaction stabilizes the ARE probe and delay the switching between the competitor and the quencher probe strand. Figure 7 compares the fluorescence intensities of the control sample and the SF-treated sample. The fluorescence intensities are different between the two samples and the assay can potentially be applied for rapid screening of other chemopreventive compounds that induce the activity of Nrf2.
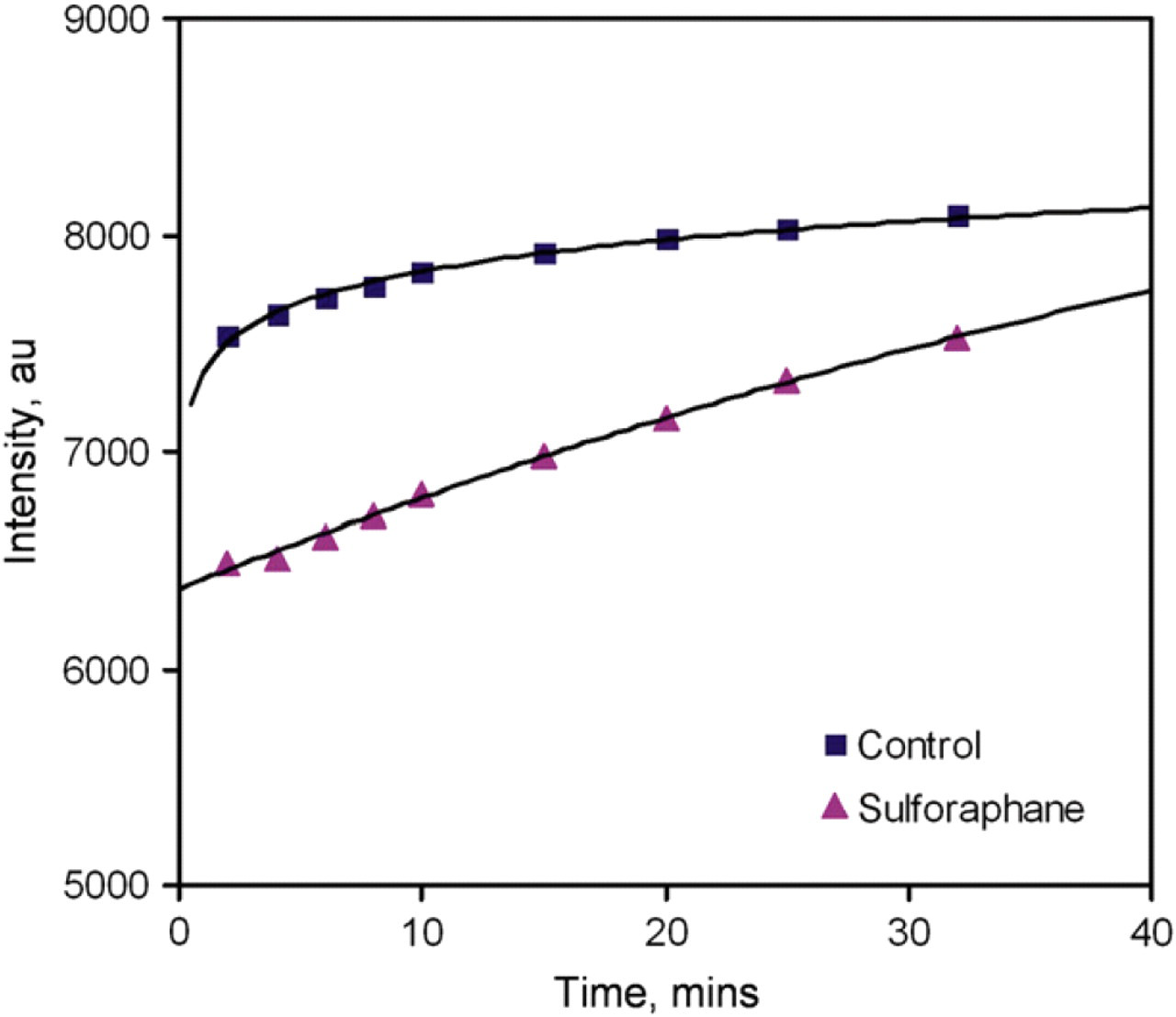
Kinetics of Nrf2 detection in whole-cell lysates. The probes consisted of 20 nM of fluorophore probe and 60 nM quencher probe. The samples were incubated with the probes for 20 min. Then, 40 nM DNA competitors were added at time zero.
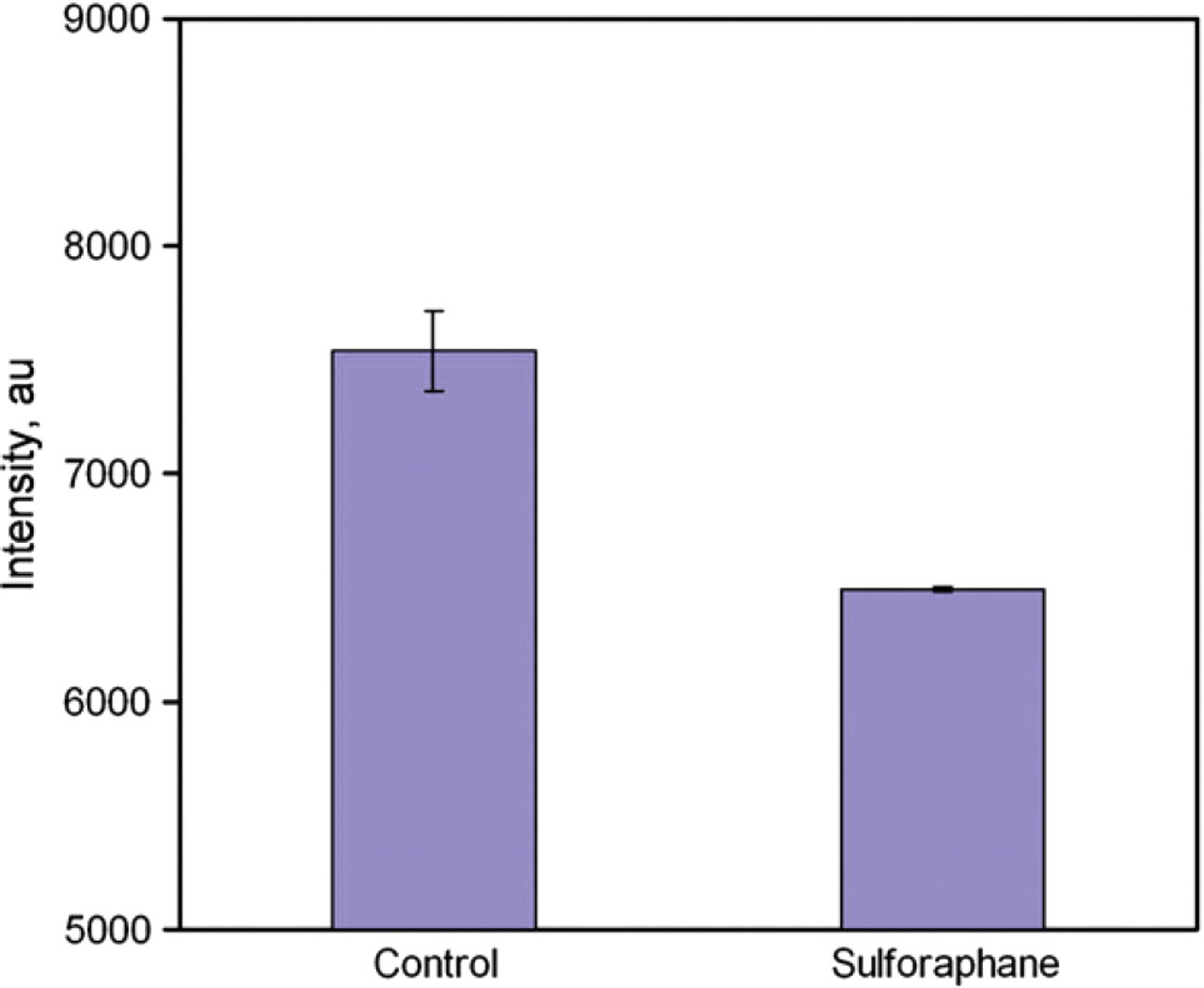
Rapid screening of Nrf2 induced by sulforaphane. Data represent mean ± standard error.
Discussion
Our experimental data demonstrated the feasibility of the molecular probe for homogeneous detection of DNA-binding proteins. Several aspects can be modified to enhance the performance of the assay. Firstly, the Nrf2 assay is based on the thermodynamic alteration as a result of the protein–DNA binding. Our data indicates that the increasing rate of the fluorescence intensity decreases with the activity of Nrf2. Nevertheless, the fluorescence intensities approach the same value for both control and SF-treated samples. It is likely that the free-energy change associated with the molecular switching is favorable even with the existence of the DNA-binding protein in the current probe design. The protein–DNA binding slows down the kinetics of the switching reaction. Therefore, a shorter competitor sequence, which is not able to switch the quencher probe strand in the existence of the DNA-binding protein, may further enhance the signal-to-noise ratio. Secondly, the experimental condition can be optimized to reduce the assay time. The assay time depends on the kinetics of the molecular switching, which is controlled by the affinity of the ARE probe. Currently, the assay can be finished in approximately 1 h. It is likely that a shorter probe sequence can further reduce the assay time. Thirdly, it is anticipated that other nonspecific DNA-binding proteins exist in the cell lysates. Nevertheless, both control cells and SF-treated cell samples showed a significant increase of fluorescence intensity. The intensity levels were similar to the control experiment without cells (not shown). It indicates only a small portion of probes are stabilized by Nrf2 or other DNA-binding proteins. It is also noteworthy that endogenous ARE exists in cell lysates. Therefore, the exogenous ARE probe is competing with endogenous ARE. At equilibrium, a portion of the Nrf2 protein (depending on the probe concentration) binds to the probe. Therefore, the probe concentration should be optimized such that a larger portion of the probes are stabilized by the protein binding. If necessary, other oligonucleotide sequences, for example, poly(dI:dC), can be introduced to prevent nonspecific binding of proteins to the ARE probe.
Conclusions
In summary, we have demonstrated the proof of principle of a molecular probe biosensor for rapid detection of Nrf2 and other DNA-binding proteins. The molecular assay can be implemented in a high-throughput microplate format or multiplexed microfluidic devices for large-scale screening of chemopreventive compounds. Further optimization of the probe design and assay conditions will improve the signal-to-noise ratio and assay time of the molecular sensor. Because a gel separation or immobilization step is not required, the equilibrium-binding kinetics of Nrf2 ARE would not be disturbed as in EMSA. More importantly, the homogenous assay allows rapid measurements of the ARE-bound Nrf2 protein and other DNA-binding protein, which is beneficial for automated drug-screening systems.
Acknowledgments
This work was supported by the Johns Hopkins Center for Alternatives to Animal Testing (2008–17) and the Arizona Cancer Center Support Grant (CA23074). Z. Wang is partially supported by the Bio5 Biology, Mathematics, and Physics Initiative (BMPI) Scholarship Award.