
Abstract

Keywords
Mass spectrometry has been described as the smallest scale in the world, not because of the mass spectrometer's size but because of the size of what it weighs—molecules. Over the past decade, mass spectrometry has undergone tremendous technological improvements, allowing for its application to proteins, peptides, carbohydrates, DNA, drugs, and many other biologically relevant molecules. Due to ionization sources such as electrospray ionization and matrix-assisted laser desorption/ionization (MALDI), mass spectrometry has become an irreplaceable tool in the biological sciences. This chapter provides an overview of mass spectrometry, focusing on ionization sources and their significance in the development of mass spectrometry in bio-molecular analysis.
A mass spectrometer determines the mass of a molecule by measuring its mass-to-charge ratio (m/z). Ions are generated by inducing either the loss or gain of a charge from a neutral species. Once formed, ions are electrostatically directed into a mass analyzer where they are separated according to m/z and finally detected. The result of molecular ionization, ion separation, and ion detection is a spectrum that can provide molecular mass or even structural information. An analogy can be drawn between a mass spectrometer and a prism, as shown in Figure 1. In the prism, light is separated into its component wavelengths, which are then detected with an optical receptor such as visualization. Similarly, in a mass spectrometer, the generated ions are separated in the mass analyzer, digitized, and detected by an ion detector (such as an electron multiplier; see chap. 2).
What is mass spectrometry?
John B. Fenn, the originator of electrospray ionization for biomolecules and the 2002 Nobel Laureate in Chemistry, probably gave the most apt answer to this question:
Mass spectrometry is the art of measuring atoms and molecules to determine their molecular weight. Such mass or weight information is sometimes sufficient, frequently necessary, and always useful in determining the identity of a species. To practice this art one puts charge on the molecules of interest, i.e., the analyte, then measures how the trajectories of the resulting ions respond in vacuum to various combinations of electric and magnetic fields. Clearly, the sine qua non of such a method is the conversion of neutral analyte molecules into ions. For small and simple species the ionization is readily carried by gas-phase encounters between the neutral molecules and electrons, photons, or other ions. In recent years, the efforts of many investigators have led to new techniques for producing ions of species too large and complex to be vaporized without substantial, even catastrophic, decomposition.
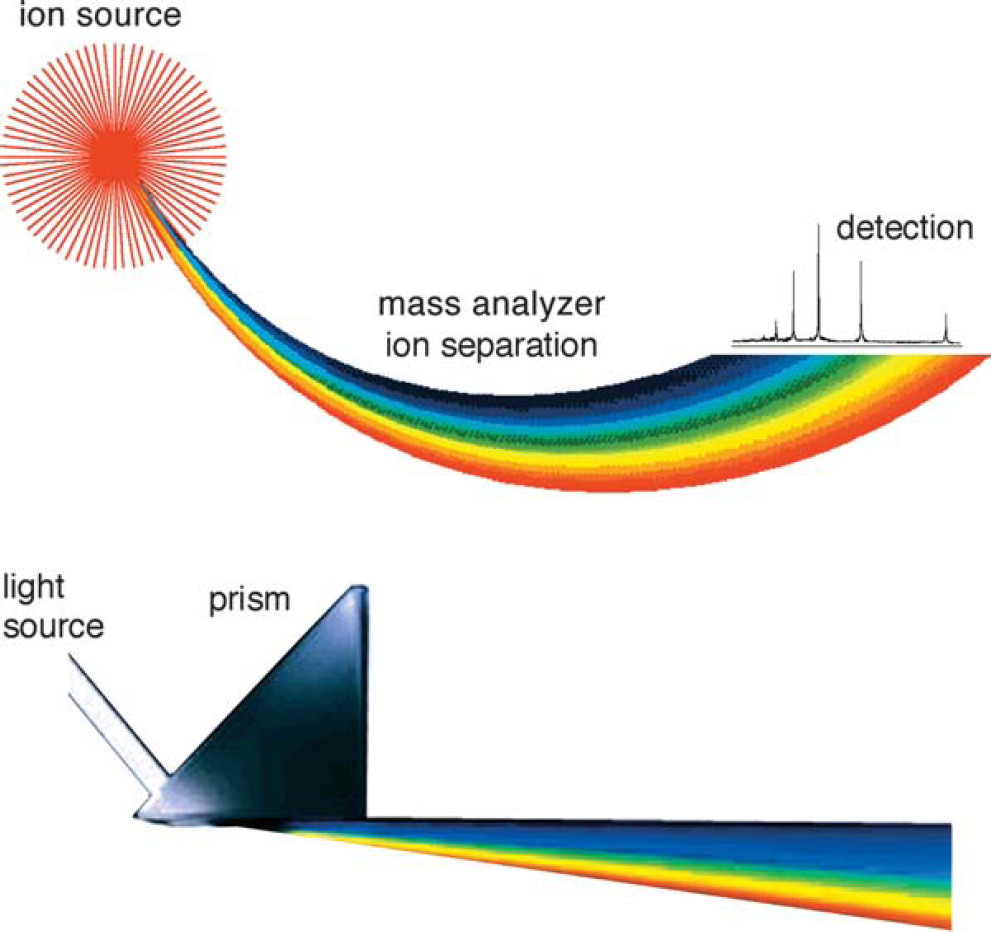
The mass analysis process as compared to the dispersion of light by a prism.
Some basics
Four basic components are, for the most part, standard in all mass spectrometers (Fig. 2): a sample inlet, an ionization source, a mass analyzer, and an ion detector. Some instruments combine the sample inlet and the ionization source, while others combine the mass analyzer and the detector. However, all sample molecules undergo the same processes regardless of instrument configuration. Sample molecules are introduced into the instrument through a sample inlet. Once inside the instrument, the sample molecules are converted to ions in the ionization source before being electrostatically propelled into the mass analyzer. Ions are then separated according to their m/z within the mass analyzer. The detector converts the ion energy into electrical signals, which are then transmitted to a computer.
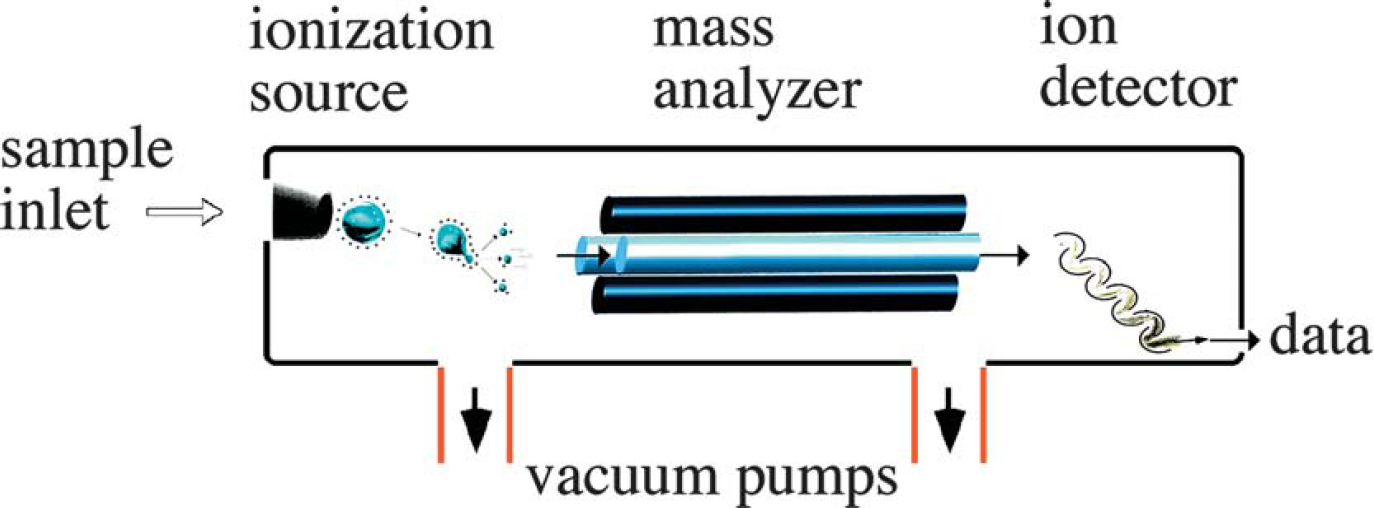
Components of a mass spectrometer. Note that the ion source does not have to be within the vacuum of the mass spectrometer. For instance, ESI and APCI are at atmospheric pressure and are known as atmospheric pressure ionization (API) sources.
Sample introduction techniques
Sample introduction was originally an important challenge in mass spectrometry. In order to perform mass analysis on a sample, which is initially at atmospheric pressure (760 Torr), it must be introduced into the instrument in such a way that the vacuum inside the instrument remains relatively unchanged (∼10–6 Torr). The most common methods of sample introduction are direct insertion with a probe or plate (commonly used with MALDI-MS), direct infusion or injection into the ionization source.
Direct Insertion: Using an insertion probe/plate (Fig. 3) is a very simple way to introduce a sample into an instrument. The sample is placed on a probe, which is then inserted into the ionization region of the mass spectrometer, typically through a vacuum interlock. The sample is either heated to facilitate thermal desorption or subjected to any number of high-energy desorption processes, such as laser desorption, to facilitate vaporization and ionization.
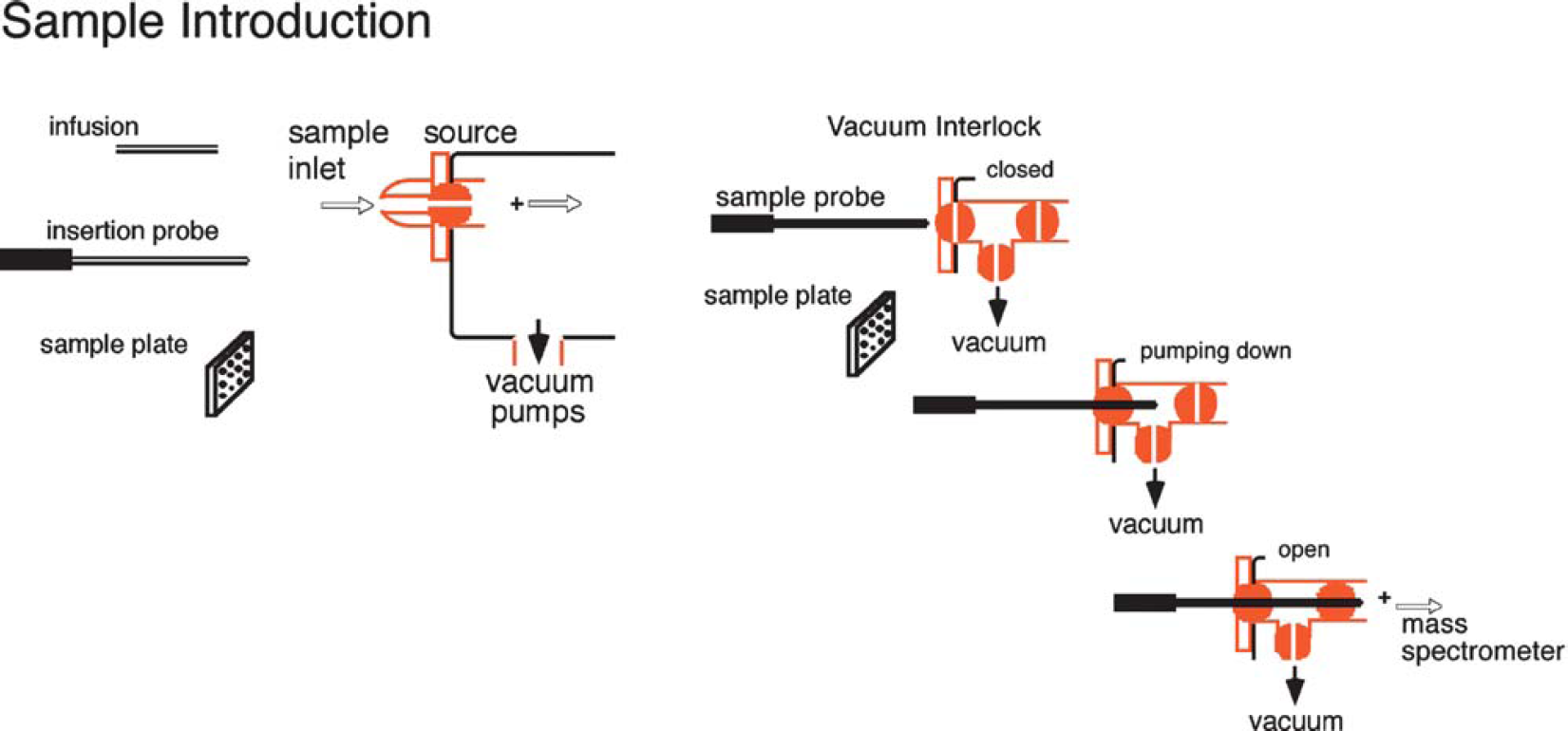
Samples are often introduced into the mass spectrometer using a direct insertion probe, a capillary column (EI with GC/MS or ESI) or a sample plate (MALDI). The vacuum interlock allows for the vacuum of the mass spectrometer to be maintained while the instrument is not in use. It also allows the sample (at atmospheric pressure) to be introduced into the high vacuum of the mass spectrometer.
Direct Infusion: A simple capillary or a capillary column is used to introduce a sample as a gas or in solution. Direct infusion is also useful because it can efficiently introduce small quantities of sample into a mass spectrometer without compromising the vacuum. Capillary columns are routinely used to interface separation techniques with the ionization source of a mass spectrometer. These techniques, including gas chromatography (GC) and liquid chromatography (LC), also serve to separate a solution's different components prior to mass analysis. In gas chromatography, separation of different components occurs within a glass capillary column. As the vaporized sample exits the gas chromatograph, it is directly introduced into the mass spectrometer.
The interface of liquid chromatography with available ionization techniques was unsuitable prior to the 1980s due to LC's low sample concentrations and relatively high flow rates. However, electrospray ionization now allows LC/MS to be performed routinely (Fig. 4). When mass spectrometry is used as a detector for these chromatographic techniques, it allows for identification of the mixture's individual components.
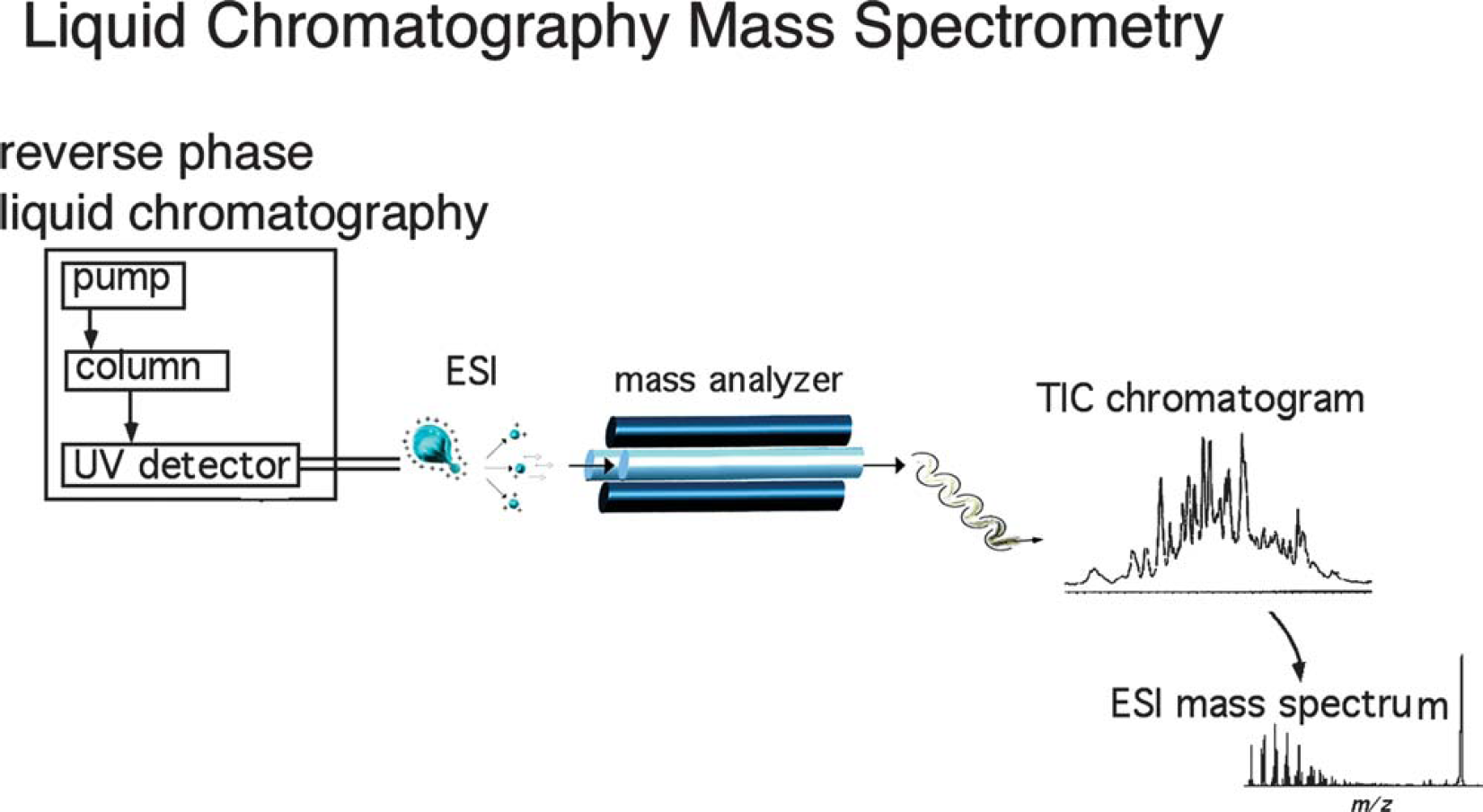
Interfacing liquid chromatography with electrospray ionization mass spectrometry. Liquid chromatography/mass spectrometry (LC/MS) ion chromatogram and the corresponding electrospray mass spectrum are shown. Gas chromatography mass spectrometry (GC/MS) produces results in much the same way as LC/MS; however, GC/MS uses an electron ionization source, which is limited by thermal vaporization (UV refers to ultraviolet and TIC is the total ion current).
Ionization
Ionization method refers to the mechanism of ionization, while the ionization source is the mechanical device that allows ionization to occur. The different ionization methods (Table 1), summarized next, work by either ionizing a neutral molecule through electron ejection, electron capture, protonation, cationization, or deprotonation, or by transferring a charged molecule from a condensed phase to the gas phase.
Ionization methods, advantages, and disadvantages

Protonation (Scheme 1) is a method of ionization by which a proton is added to a molecule, producing a net positive charge of 1+ for every proton added. Positive charges tend to reside on the more basic residues of the molecule, such as amines, to form stable cations. Peptides are often ionized via protonation. Protonation can be achieved via MALDI, electrospray ionization (ESI), and atmospheric pressure chemical ionization (APCI).
An example of a mass spectrum obtained via protonation.
Deprotonation (Scheme 2) is an ionization method by which the net negative charge of 1- is achieved through the removal of a proton from a molecule. This mechanism of ionization, commonly achieved via MALDI, ESI, and APCI, is very useful for acidic species, including phenols, carboxylic acids, and sulfonic acids. The negative ion mass spectrum of sialic acid is shown in Scheme 2.
An example of a mass spectrum of sialic acid obtained via deprotonation.
Cationization (Scheme 3) is a method of ionization that produces a charged complex by noncovalently adding a positively charged ion to a neutral molecule. While protonation could fall under this same definition, cationization is distinct for its addition of a cation adduct other than a proton (e.g., alkali, ammonium). Moreover, it is known to be useful with molecules unstable to protonation. The binding of cations other than protons to a molecule is naturally less covalent; therefore, the charge remains localized on the cation. This minimizes delocalization of the charge and fragmentation of the molecule. Cationization is commonly achieved via MALDI, ESI, and APCI. Carbohydrates are excellent candidates for this ionization mechanism, with Na+ a common cation adduct.
An example of a mass spectrum obtained via cationization.
The transfer (Scheme 4) of compounds already charged in solution is normally achieved through the desorption or ejection of the charged species from the condensed phase into the gas phase. This transfer is commonly achieved via MALDI or ESI. The positive ion mass spectrum of tetraphenylphosphine is shown in Scheme 4.

An example of a mass spectrum of tetraphenylphosphine obtained via transfer of a charged species from solution into the gas phase.
As its name implies, electron ejection (Scheme 5) achieves ionization through the ejection of an electron to produce a 1+ net positive charge, often forming radical cations. Observed most commonly with electron ionization (EI) sources, electron ejection is usually performed on relatively nonpolar compounds with low molecular weights and is known to generate significant fragment ions. The mass spectrum resulting from electron ejection of anthracene is shown in Scheme 5.
An example of a mass spectrum obtained via electron ejection.
With the electron capture ionization method (Scheme 6), a net negative charge of 1- is achieved with the absorption or capture of an electron. It is a mechanism of ionization primarily observed for molecules with a high electron affinity such as halogenated compounds. The electron capture mass spectrum of hexachlorobenzene is shown in Scheme 6.
An example of a mass spectrum obtained via electron capture. Electron capture is commonly achieved via EI.
Ionization sources
Prior to the 1980s, EI was the primary ionization source for mass analysis. However, EI limited chemists and biochemists to small molecules well below the mass range of common bioorganic compounds. This limitation motivated scientists such as John B. Fenn, Koichi Tanaka, Franz Hillenkamp, Michael Karas, Graham Cooks, and Michael Barber to develop the new generation of ionization techniques, including fast atom/ion bombardment (FAB), MALDI, and ESI (Table 2). These techniques have revolutionized biomolecular analyses, especially for large molecules. Among them, ESI and MALDI have clearly evolved to be the methods of choice when it comes to biomolecular analysis.
Summary of ionization sources, their acronyms, and method of ionization

MALDI and ESI are now the most common ionization sources for biomolecular mass spectrometry, offering excellent mass range and sensitivity (Fig. 5). The following section focuses on the principles of ionization sources, providing some details on the practical aspects of their use as well as ionization mechanisms.
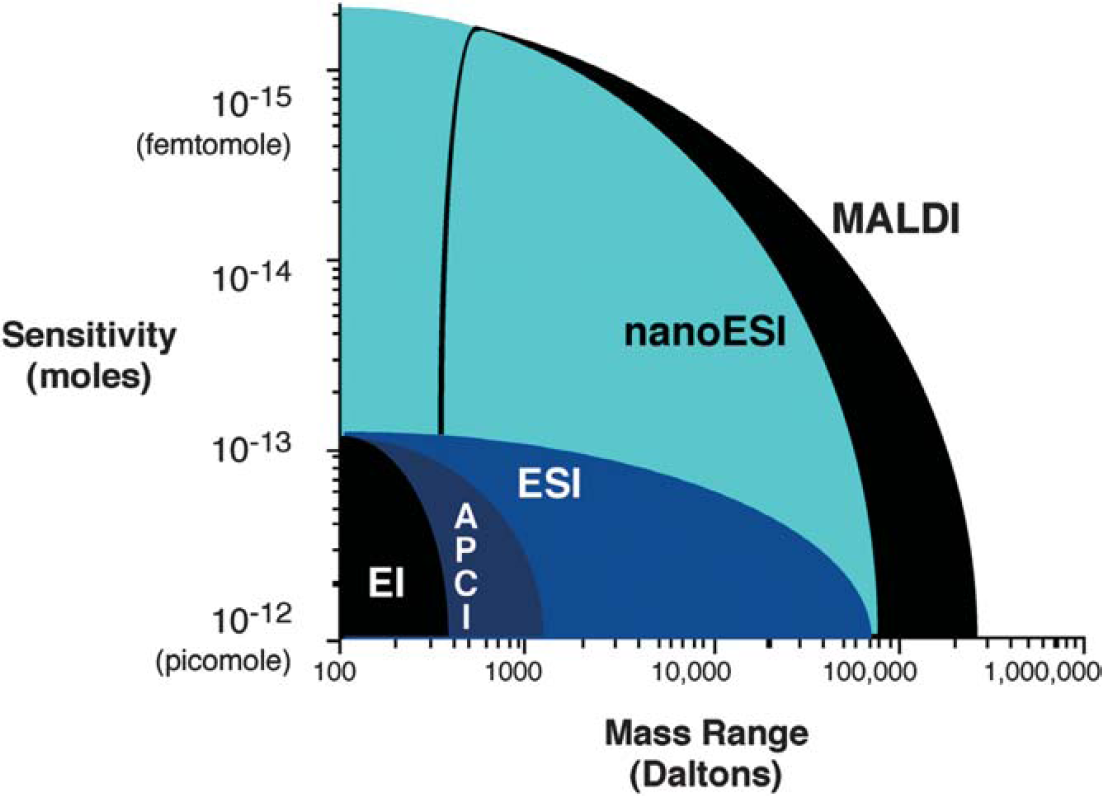
A glance at the typical sensitivity and mass ranges allowed by different ionization techniques provides a clear answer to the question of which are most useful; EI, APCI, and DIOS are somewhat limiting in terms of upper mass range, while ESI, nanoESI, and MALDI have a high practical mass range.
Electrospray Ionization
The idea of electrospray, while not new, has been rejuvenated with its recent application to biomolecules. The first electrospray experiments were carried out by Chapman in the late 1930s, and the practical development of electro-spray sample ionization for mass spectrometry was accomplished by Dole in the late 1960s. Dole also discovered the important phenomenon of multiple charging of molecules. Fenn's work ultimately led to the modern-day technique of electrospray ionization mass spectrometry and its application to biological macromolecules.
ESI is a method routinely used with peptides, proteins, carbohydrates, small oligonucleotides, synthetic polymers, and lipids (Table 3). ESI produces gaseous ionized molecules directly from a liquid solution. It operates by creating a fine spray of highly charged droplets in the presence of an electric field. (An illustration of the electrospray ionization process is shown in Figs. 6 and 7). The sample solution is sprayed from a region of the strong electric field at the tip of a metal nozzle maintained at a potential of anywhere from 700 to 5000 V. The nozzle (or needle) to which the potential is applied serves to disperse the solution into a fine spray of charged droplets. Dry gas, heat, or both are applied to the droplets at atmospheric pressure, thus causing the solvent to evaporate from each droplet. As the size of the charged droplet decreases, the charge density on its surface increases. The mutual Coulombic repulsion between like charges on this surface becomes so great that it exceeds the forces of surface tension, and ions are ejected from the droplet through what is known as a “Taylor cone.” Another possibility is that the droplet explodes, releasing the ions. In either case, the emerging ions are directed into an orifice through electrostatic lenses leading to the vacuum of the mass analyzer. Because ESI involves the continuous introduction of solution, it is suitable for using as an interface with high-performance liquid chromatography (HPLC) or capillary electrophoresis.
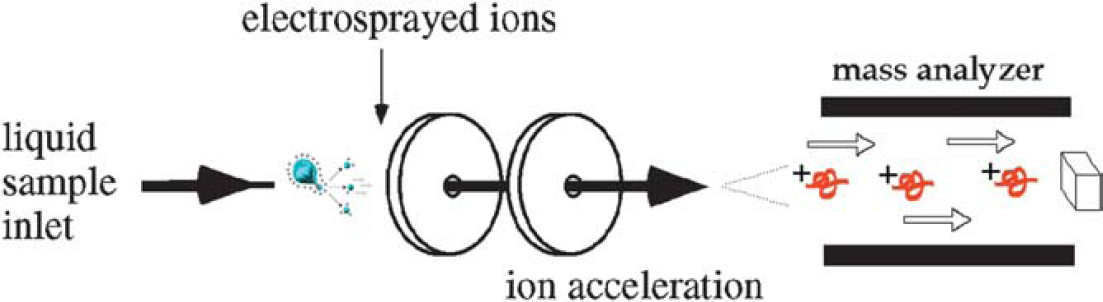
ESI mass spectrometry.
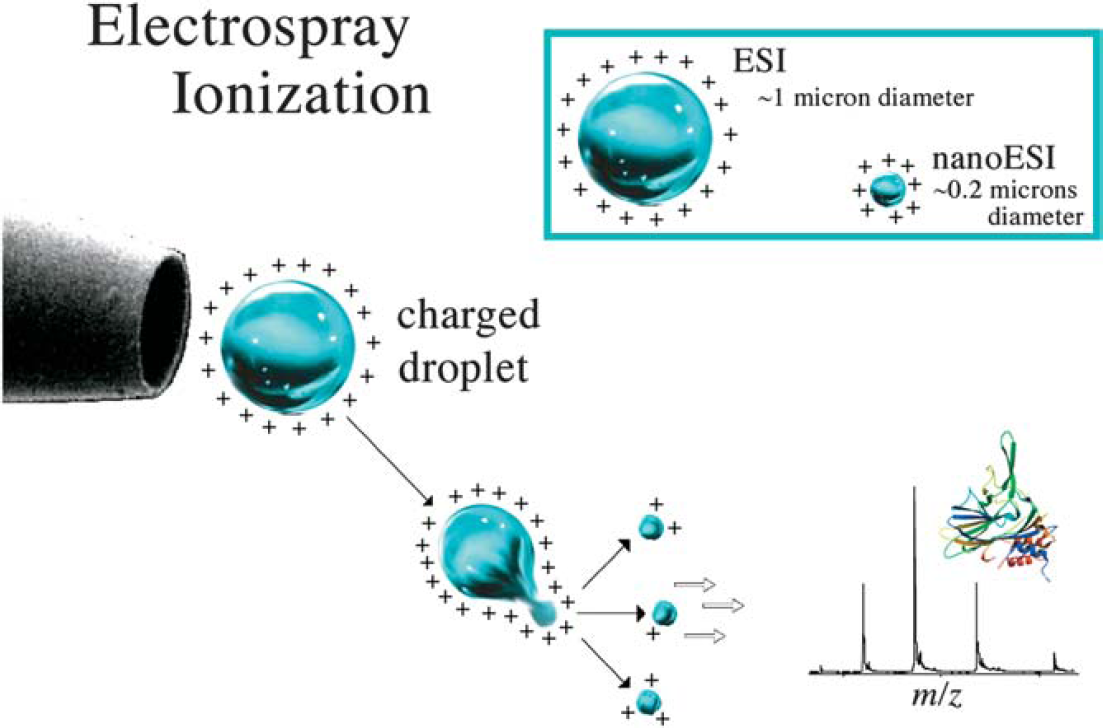
Ion formation from electrospray ionization source. The electrospray ionization source uses a stream of air or nitrogen, heat, a vacuum, or a solvent sheath (often methanol) to facilitate desolvation of the droplets. After the ions form (at atmospheric pressure), they are electrostatically directed into the mass analyzer.
Advantages and disadvantages of electrospray ionization (ESI)

Electrospray ionization is conducive to the formation of singly charged small molecules but is also well known for producing multiply charged species of larger molecules. This is an important phenomenon because the mass spectrometer measures the m/z and, therefore, multiple charging makes it possible to observe very large molecules with an instrument having a relatively small mass range. Fortunately, software available with all electrospray mass spectrometers facilitates the molecular weight calculations necessary to determine the mass of the multiply charged species. Figures 8 and 9 illustrate the different charge states on two different proteins, where each of the peaks in the mass spectra can be associated with different charge states of the molecular ion. Multiple charging has other important advantages in tandem mass spectrometry. One advantage is that upon fragmentation, you observe more fragment ions with multiply charged precursor ions than with singly charged precursor ions.
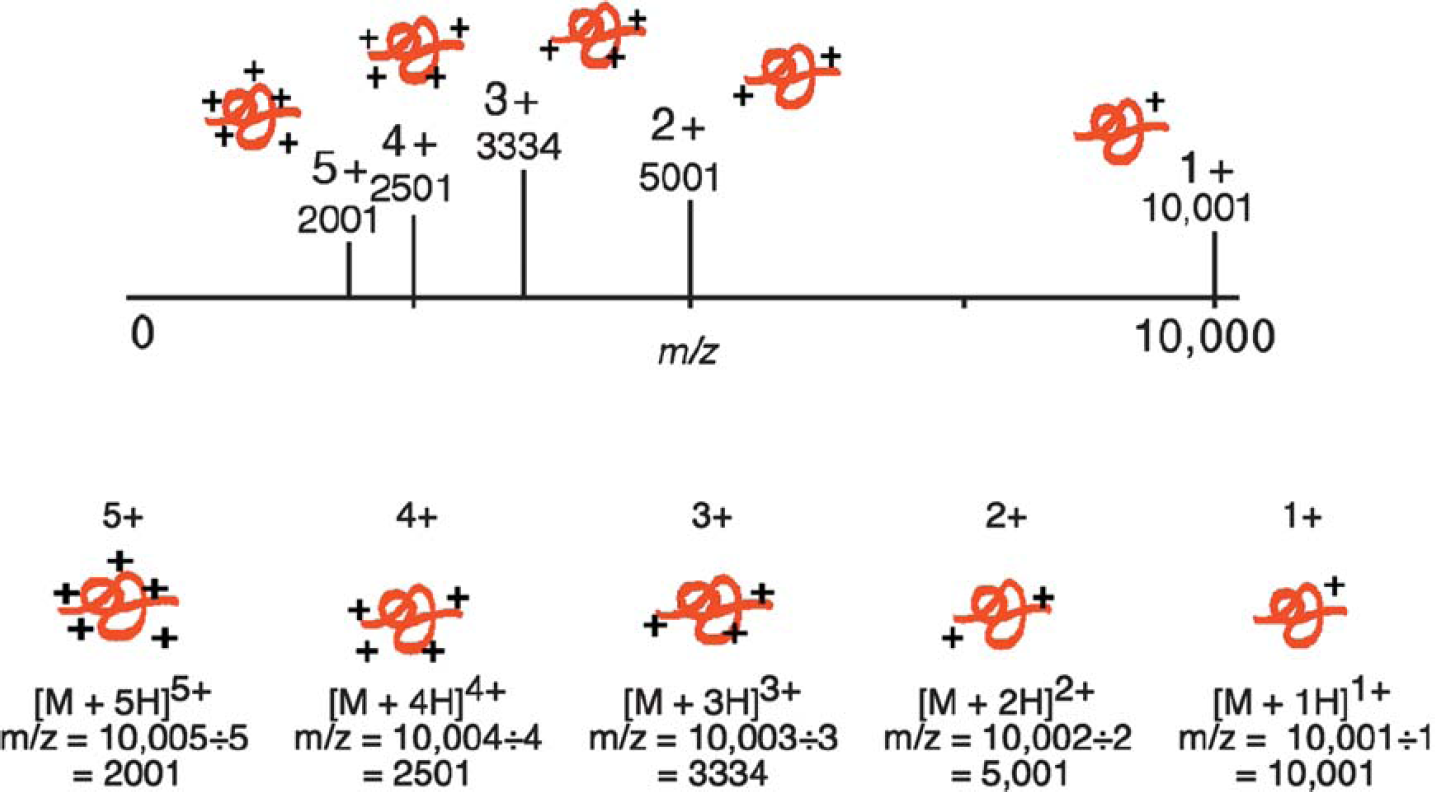
A protein with a molecular weight of 10,000 generates five different peaks with the ions containing 5, 4, 3, 2, and 1 charges, respectively. The mass spectrometer detects each of the protein ions at 2001, 2501, 3334, 5001, and 10,001, respectively.
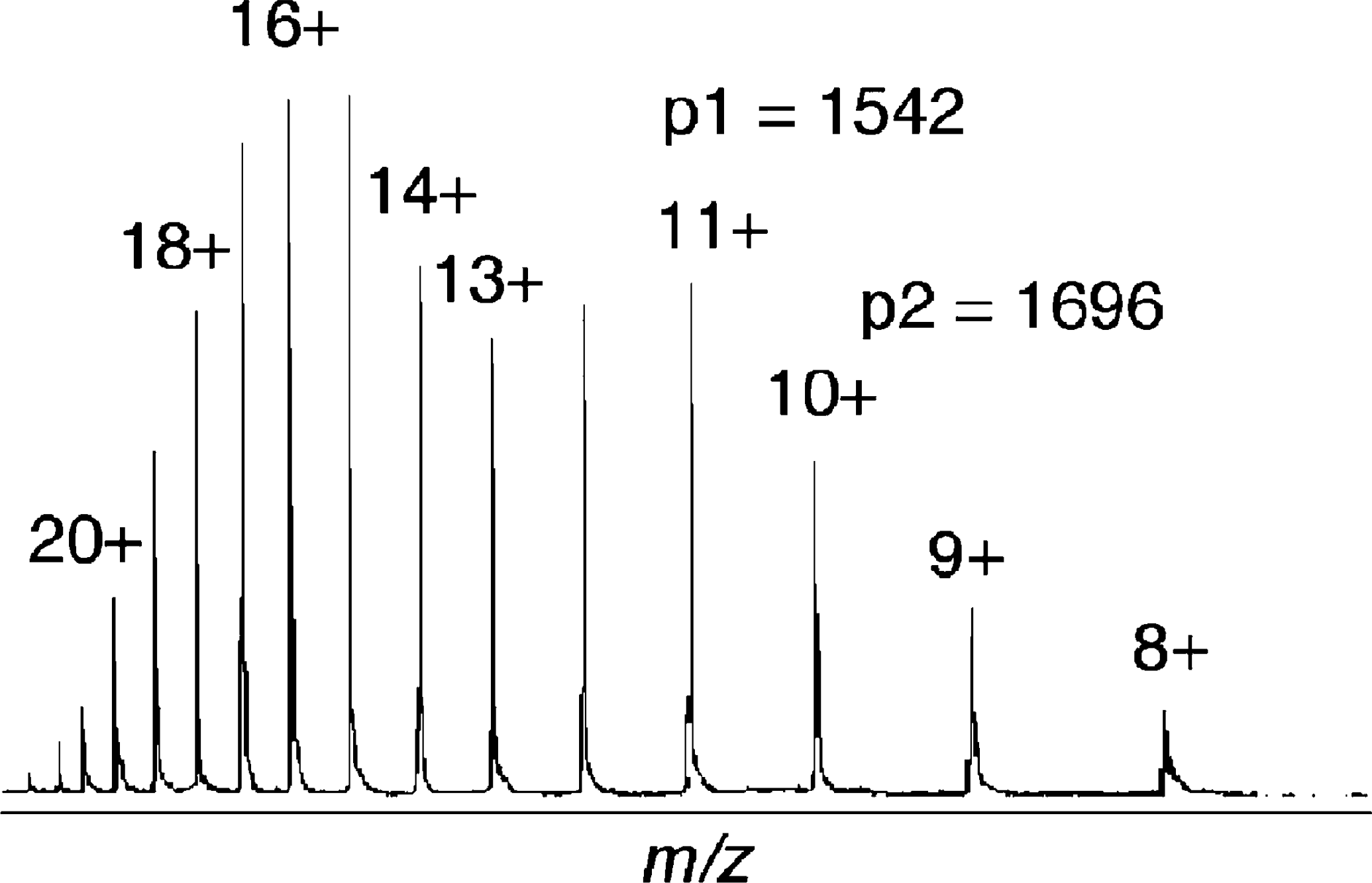
The multiply charged ions of myoglobin generated from ESI. The different peaks represent different charge states of myoglobin. The molecular weight can be determined using Eqn. 1-3.
Multiple charging: A 10,000-Da protein and its theoretical mass spectrum with up to five charges are shown in Figure 8. The mass of the protein remains the same, yet the m/z varies depending upon the number of charges on the protein. Protein ionization is usually the result of protonation, which not only adds charge but also increases the mass of the protein by the number of protons added. This effect on the m/z applies equally for any mechanism of molecular ionization resulting in a positively or negatively charged molecular ion, including the addition or ejection of charge-carrying species other than protons (e.g., Na+ and Cs+). Multiple positive charges are observed for proteins, while for oligonucleotides, negative charging (with ESI) is typical.
Although electrospray mass spectrometers are equipped with software that can calculate molecular weight, an understanding of how the computer makes such calculations from multiply charged ions is beneficial. Eqn. 1 to 5 and Figure 9 offer a simple explanation, where we assume p1 and p2 are adjacent peaks and differ by a single charge, which is equivalent to the addition of a single proton.


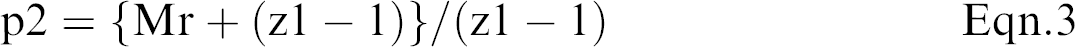
p = a peak in the mass spectrum
m = total mass of an ion
z = total charge
Mr = average mass of protein
p1 = m/z value for p1
p2 = m/z value for p2
z1 = charge on peak p1
Eqn. 2 and 3 can be solved for the two unknowns, Mr and z1.
For the peaks in the mass spectrum of myoglobin shown in Figure 9, p1 = 1542, and p2 = 1696.

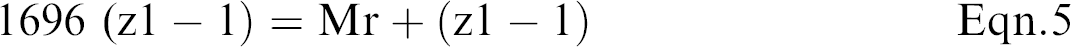
Solving the two equations: Mr = 16,951 Da for z1 = 11.
Electrospray Solvents
Many solvents can be used in ESI and are chosen based on the solubility of the compound of interest, the volatility of the solvent, and the solvent's ability to donate a proton. Typically, protic primary solvents such as methanol, 50/50 methanol/water, or 50/50 acetonitrile/H2O are used, while aprotic cosolvents, such as 10% DMSO in water as well as isopropyl alcohol, are used to improve solubility for some compounds. Although 100% water is used in ESI, water's relatively low vapor pressure has a detrimental effect on sensitivity; better sensitivity is obtained when a volatile organic solvent is added. Some compounds require the use of straight chloroform with 0.1% formic acid added to facilitate ionization. This approach, while less sensitive, can be effective for otherwise insoluble compounds. Buffers such as Na+, K+ phosphate, and salts present a problem for ESI by lowering the vapor pressure of the droplets, and they significantly reduce signal in ESI through reduction of volatility. Consequently, volatile buffers such as ammonium acetate can be used more effectively.
Configuration of the Electrospray Ion Source
The orthogonal ESI configuration now used in many instruments to introduce the ions into the analyzers (as shown in Fig. 10) has turned out to be very valuable for high flow rate applications. The primary advantage of this configuration is that the flow rates can be increased without contaminating or clogging the inlet. Orthogonal spraying is important because the entrance to the analyzer is no longer being saturated by solvent, thus keeping droplets from entering and contaminating the inlet. Instead, only ions are directed toward the inlet. This makes ESI even more compatible with LC/MS at the milliliter-per-minute flow rates.

An example of orthogonal ESI.
Nanoelectrospray Ionization (NanoESI)
Low flow electrospray, originally described by Wilm and Mann, has been called nanoelectrospray, nanospray, and microelectrospray. This ionization source is a variation on ESI, where the spray needle has been made very small and is positioned close to the entrance to the mass analyzer. The end-result of this rather simple adjustment is increased efficiency, which includes a reduction in the amount of sample needed.
The flow rates for nanoESI sources are on the order of tens to hundreds of nanoliters per minute. In order to obtain these low flow rates, nanoESI uses emitters of pulled and, in some cases, metallized glass that have a small orifice (∼5 μm). Also, the emitters are positioned very close to the entrance of the mass analyzer; therefore, ion transmission to the mass analyzer is much more efficient and sensitive. For instance, the analysis of a 5-μM solution of a peptide by nanoESI would be performed in 1 min, consuming ∼50 femtomoles of sample. The same experiment performed with normal ESI in the same time would require 5 picomoles, or 100 times more sample than for nanoESI. In addition, since the droplets are typically smaller with nanoESI than normal ESI, the amount of evaporation necessary to obtain ion formation is much less. Consequently, NanoESI is more tolerant of salts and other impurities because less evaporation means the impurities are not concentrated down as much as they are in ESI.
Atmospheric Pressure Chemical Ionization (APCI)
APCI has also become an important ionization source because it generates ions directly from solution and is capable of analyzing relatively nonpolar compounds. Similar to electrospray, the liquid effluent of APCI (Fig. 11) is introduced directly into the ionization source. However, the similarity stops there. The droplets are not charged, and the APCI source contains a heated vaporizer, which facilitates rapid desolvation/vaporization of the droplets. Vaporized sample molecules are carried through an ion-molecule reaction region at atmospheric pressure. The ionization originates from the solvent being excited/ionized from the corona discharge. Because the solvent ions are present at atmospheric pressure conditions, chemical ionization of analyte molecules is very efficient; at atmospheric pressure, analyte molecules collide with the reagent ions frequently. Proton transfer (for protonation MH+ reactions) occurs in the positive mode and either electron transfer or proton loss ([M-H]-) in the negative mode. The moderating influence of the solvent clusters on the reagent ions, and of the high gas pressure, reduces fragmentation during ionization and results in primarily intact molecular ions. Multiple charging is typically not observed, presumably because the ionization process is more energetic than ESI.
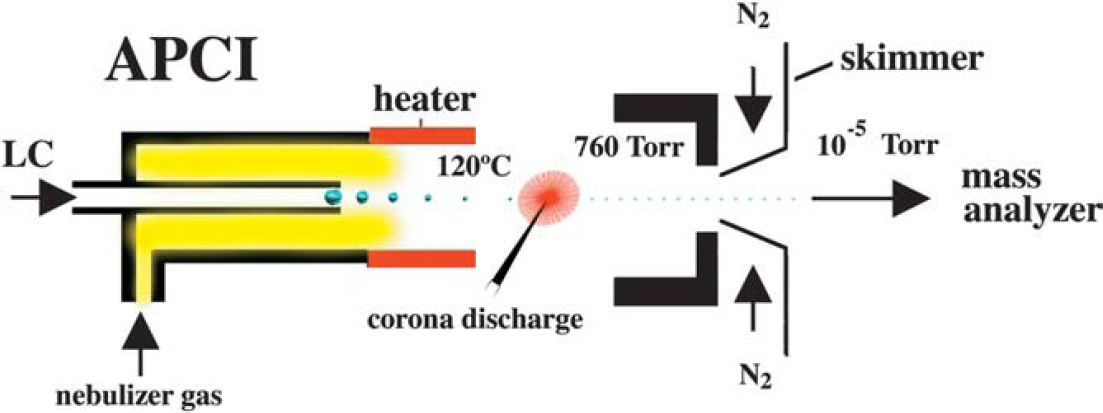
APCI mass spectrometry.
Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry (MALDI-MS)
MALDI-MS (Table 4) was first introduced in 1988 by Tanaka, Karas, and Hillenkamp. It has since become a widespread analytical tool for peptides, proteins, and most other biomolecules (oligonucleotides, carbohydrates, natural products, and lipids). The efficient and directed energy transfer during a matrix-assisted laser-induced desorption event provides high ion yields of the intact analyte and allows for the measurement of compounds with sub-picomole sensitivity. In addition, the utility of MALDI for the analysis of heterogeneous samples makes it very attractive for the mass analysis of complex biological samples such as proteolytic digests.
Advantages and disadvantages of matrix-assisted laser desorption/ionization (MALDI)

While the exact desorption/ionization mechanism for MALDI is not known, it is generally believed that MALDI (Fig. 12) causes the ionization and transfer of a sample from the condensed phase to the gas phase via laser excitation and vaporization of the sample matrix. In MALDI analysis, the analyte is first co-crystallized with a large molar excess of a matrix compound (Figs. 13 and 14), usually a UV-absorbing weak organic acid. Irradiation of this analytematrix mixture by a laser results in the vaporization of the matrix, which carries the analyte with it. The matrix plays a key role in this technique. The co-crystallized sample molecules also vaporize, but without having to directly absorb energy from the laser. Molecules sensitive to the laser light are therefore protected from direct UV laser excitation.

The efficient and directed energy transfer of the UV laser pulse during a MALDI event allows for relatively small quantities of sample (femtomole to picomole) to be analyzed. In addition, the utility of MALDI mass spectrometry for the analysis of heterogeneous samples makes it very attractive for the mass analysis of biological samples.
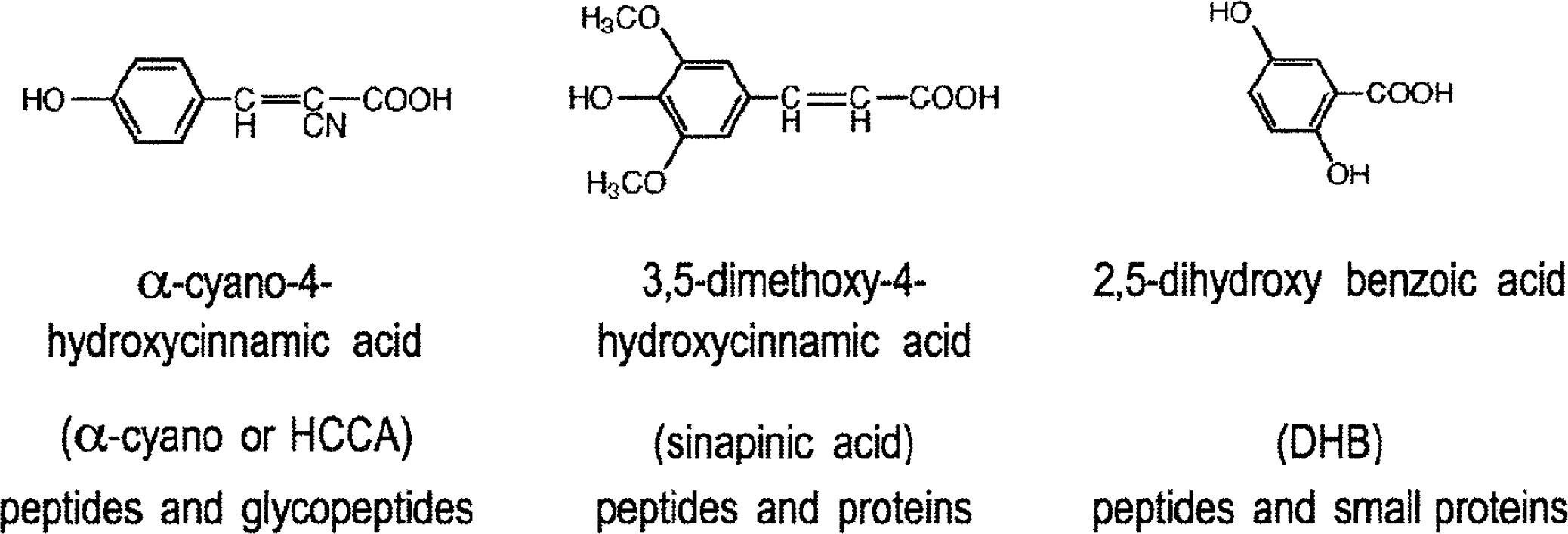
Commonly used MALDI matrices.
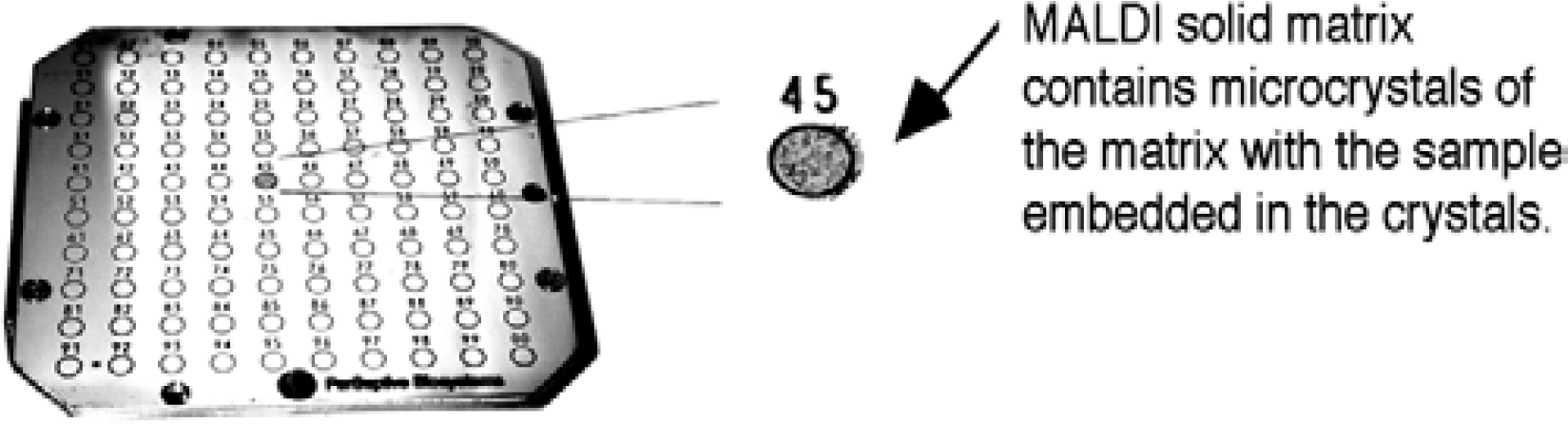
One of the advantages of MALDI is that multiple samples can be prepared at the same time, as seen with this multisample plate containing 100 positions.
MALDI matrix: A nonvolatile solid material facilitates the desorption and ionization process by absorbing the laser radiation. As a result, both the matrix and any sample embedded in the matrix are vaporized. The matrix also serves to minimize sample damage from laser radiation by absorbing most of the incident energy.
Once in the gas phase, the desorbed charged molecules are then directed electrostatically from the MALDI ionization source (Table 5) into the mass analyzer. Time-of-flight (TOF) mass analyzers are often used to separate the ions according to their m/z. The pulsed nature of MALDI is directly applicable to TOF analyzers since the ion's initial TOF can be started with each pulse of the laser and completed when the ion reaches the detector.
General comparison of ionization sources

Several theories have been developed to explain desorption by MALDI. The thermal-spike model proposes that the ejection of intact molecules is attributed to poor vibrational coupling between the matrix and analyte, which minimizes vibrational energy transfer from the matrix to the vibrational modes of the analyte molecule, thereby minimizing fragmentation. The pressure pulse theory proposes that a pressure gradient from the matrix is created normal to the surface and desorption of large molecules is enhanced by momentum transfer from collisions with these fast-moving matrix molecules. It is generally thought that ionization occurs through proton transfer or cationization during the desorption process.
The utility of MALDI for biomolecule analyses lies in its ability to provide molecular weight information on intact molecules. The ability to generate accurate information can be extremely useful for protein identification and characterization. For example, a protein can often be unambiguously identified by the accurate mass analysis of its constituent peptides (produced by either chemical or enzymatic treatment of the sample).
Sample-matrix preparation procedures greatly influence the quality of MALDI mass spectra of peptides/proteins. Among the variety of reported preparation methods, the dried-droplet method is the most frequently used. In this case, a saturated matrix solution is mixed with the analyte solution, giving a matrix-to-sample ratio of about 5000:1. An aliquot (0.5-2.0 μL) of this mixture is then applied to the sample target where it is allowed to dry. Below is an example of how the dried-droplet method is performed:
Pipet 0.5 μL of sample to the sample plate.
Pipet 0.5 μL of matrix to the sample plate.
Mix the sample and matrix by drawing the combined droplet in and out of the pipette.
Allow to air dry.
For peptides, small proteins and most compounds: A saturated solution of α-cyano-4-hydroxycinnamic acid in 50:50 ACN:H2O with 0.1% TFA.
For proteins and other large molecules: A saturated solution of sinapinic acid in 50:50 ACN:H2O with 0.1% TFA.
For glycopeptides/proteins and small compounds: A saturated solution of 2,5-dihydroxy benzoic acid (DHB) in 50:50 ACN:H2O.
Alternatively, samples can be prepared in a stepwise manner. In the thin-layer method, a homogeneous matrix “film” is formed on the target first, and the sample is then applied and absorbed by the matrix. This method yields good sensitivity, resolving power, and mass accuracy. Similarly, in the thick-layer method, nitrocellulose (NC) is used as the matrix additive; once a uniform NC-matrix layer is obtained on the target, the sample is applied. This preparation method suppresses alkali adduct formation and significantly increases the detection sensitivity, especially for peptides and proteins extracted from gels. The sandwich method is another variant in this category. A thin layer of matrix crystals is prepared as in the thin-layer method, followed by the subsequent addition of droplets of (a) aqueous TFA, (b) sample, and (c) matrix.
Desorption/Ionization on Silicon (DIOS)
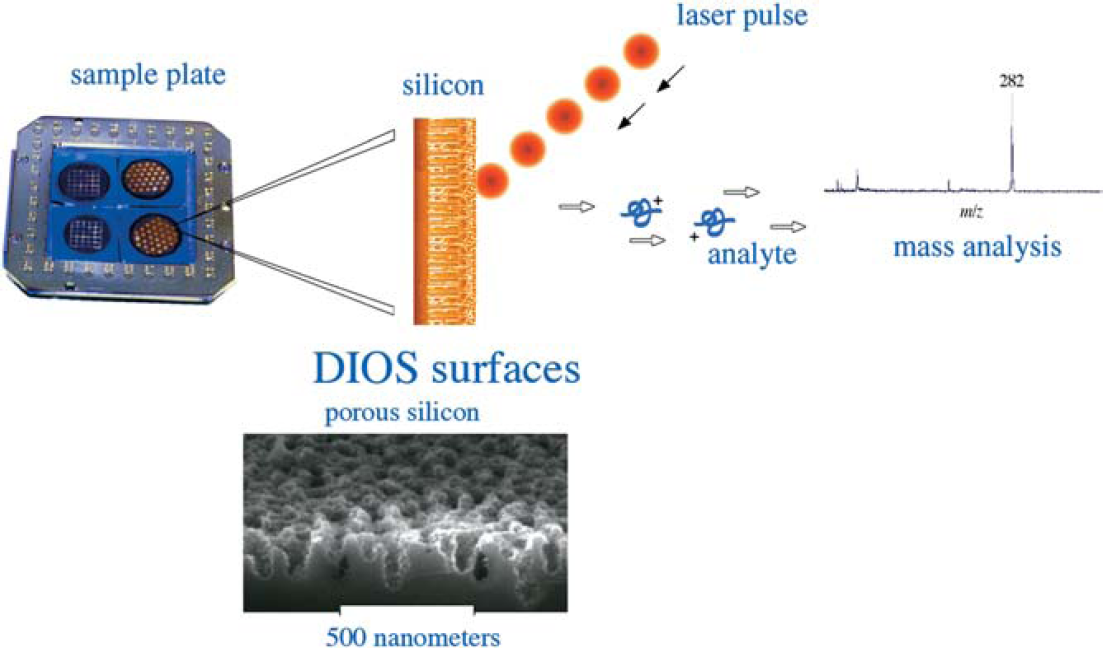
DIOS is a matrix-free method that uses pulsed laser desorption/ionization on porous silicon (Fig. 15). Porous silicon is a UV-absorbing semiconductor with a large surface area (hundreds of m2/cm3) and is produced through electrochemical anodization or chemical etching of crystalline silicon. Much of the interest in porous silicon and its morphology derives from its photoluminescent properties, which make it a useful platform for electronic and optoelectronic devices as well as chemical microsensors. For its application to laser desorption/ionization mass spectrometry, the structure of porous silicon provides a scaffold for retaining solvent and analyte molecules, and the UV absorptivity affords a mechanism for the transfer of the laser energy to the analyte. This fortuitous combination of characteristics allows DIOS to be useful for a large variety of biomolecules, including peptides, carbohydrates, and small organic compounds of various types. Unlike other direct, matrix-free desorption techniques, DIOS enables desorption/ionization with little or no analyte degradation.
The efficient and directed energy transfer of the UV laser pulse during a DIOS event allows for relatively small quantities of sample (femtomole to picomole) to be analyzed. In addition, DIOS mass spectrometry is useful for the analysis of heterogeneous samples and small molecules. On the left is a picture of a DIOS chip; the dark spots represent porous silicon. On the right is a diagrammatic representation of the DIOS event from a chip.
DIOS has a great deal in common with MALDI. Instrumentation and acquisition using DIOS-MS requires only minor adjustments to the MALDI setup—the chips are simply affixed to a machined MALDI plate and inserted into the spectrometer. The same wavelength of laser light (337 nm) typically employed in MALDI is effective for DIOS. While DIOS is comparable to MALDI with respect to its sensitivity, it has several advantages due to the lack of interfering matrix: low background in the low mass range, uniform deposition of aqueous samples, and simplified sample handling. In addition, the chip-based format can be adapted to automated sample handling, where the laser rapidly scans from spot to spot. DIOS could thus accelerate and simplify high-throughput analysis of low molecular weight compounds, as MALDI has done for macromolecules. Because the masses of many low molecular weight compounds can be measured, DIOS-MS can be applied to the analysis of small molecule transformations, both enzymatic and chemical.
Fast Atom/Ion Bombardment
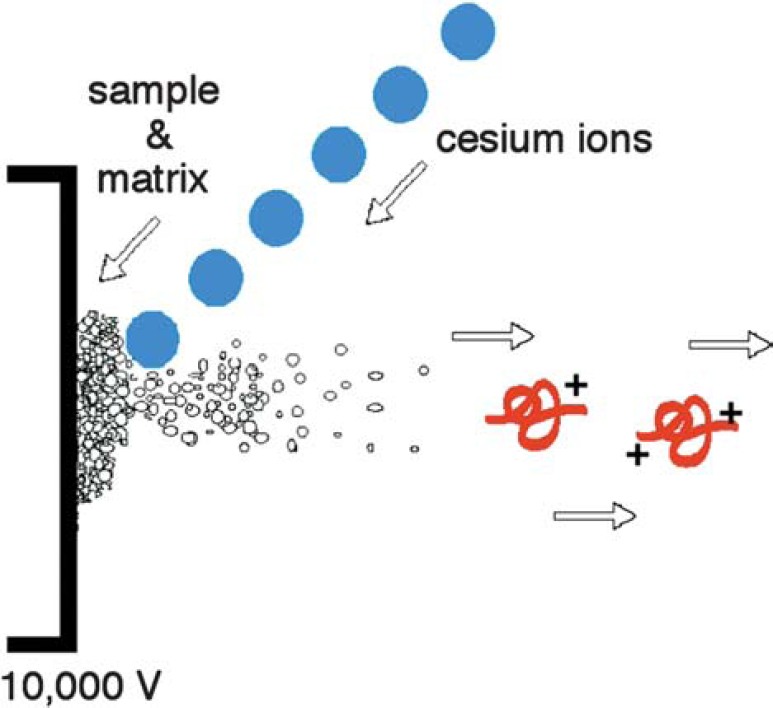
Fast atom ion bombardment, or FAB (Fig. 16), is an ionization source similar to MALDI in that it uses a matrix and a highly energetic beam of particles to desorb ions from a surface. It is important, however, to point out the differences between MALDI and FAB. For MALDI, the energy beam is pulsed laser light, while FAB uses a continuous ion beam. With MALDI, the matrix is typically a solid crystalline, whereas FAB typically has a liquid matrix. It is also important to note that FAB is about 1000 times less sensitive than MALDI.
FAB mass spectrometry, also known as liquid secondary ion mass spectrometry (LSIMS).
Fast atom bombardment is a soft ionization source that requires the use of a direct insertion probe for sample introduction and a beam of Xe neutral atoms or Cs+ ions to sputter the sample and matrix from the direct insertion probe surface. It is common to detect matrix ions in the FAB spectrum as well as the protonated or cationized (i.e., M+Na+) molecular ion of the analyte of interest.
FAB matrix: Facilitating the desorption and ionization process, the FAB matrix is a nonvolatile liquid material that serves to constantly replenish the surface with new sample as it is bombarded by the incident ion beam. By absorbing most of the incident energy, the matrix also minimizes sample degradation from the high-energy particle beam.
Two of the most common matrices used with FAB are m-nitrobenzyl alcohol and glycerol (Fig. 17).
Two common matrices for fast atom bombardment (FAB).
The fast atoms or ions impinge on or collide with the matrix causing the matrix and analyte to be desorbed into the gas phase. The sample may already be charged and get transferred into the gas phase by FAB, or it may become charged during FAB desorption through reactions with surrounding molecules or ions. Once in the gas phase, the charged molecules can be propelled electrostatically to the mass analyzer.
Electron Ionization (EI)
Electron ionization is one of the most important ionization sources for the routine analysis of small, hydrophobic, thermally stable molecules and is still widely used. Because EI usually generates numerous fragment ions, it is a “hard” ionization source. However, the fragmentation information can also be very useful. For example, by employing databases containing over 200,000 electron ionization mass spectra, it is possible to identify an unknown compound in seconds (provided it exists in the database). These databases, combined with current computer storage capacity and searching algorithms, allow for rapid comparison with these databases (such as the NIST database), thus greatly facilitating the identification of small molecules.
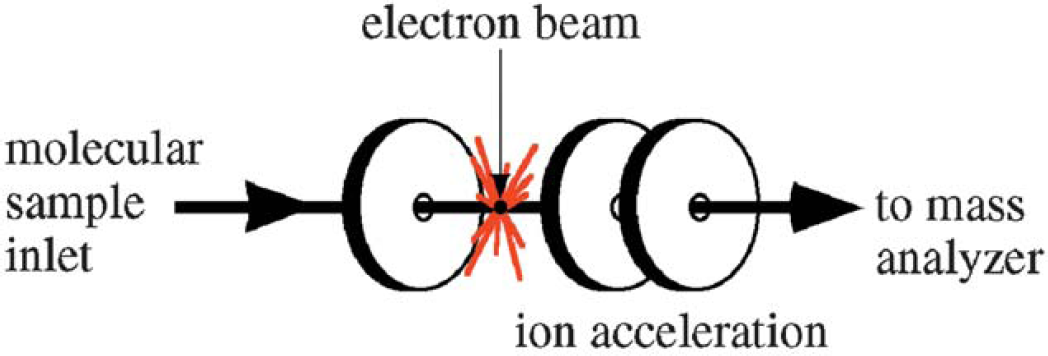
The electron ionization source is straightforward in design. The sample must be delivered as a gas which is accomplished by either “boiling off” the sample from a probe via thermal desorption or by introduction of a gas through a capillary. The capillary is often the output of a capillary column from gas chromatography instrumentation. In this case, the capillary column provides separation (this is also known as gas chromatography mass spectrometry or GC/MS). Desorption of both solid and liquid samples is facilitated by heat as well as the vacuum of the mass spectrometer. Once in the gas phase, the compound passes into an electron ionization source (Fig. 18), where electrons excite the molecule, thus causing electron ejection ionization and fragmentation.
EI mass spectrometry.
The utility of electron ionization decreases significantly for compounds above a molecular weight of 400 Da because the required thermal desorption of the sample often leads to thermal decomposition before vaporization is able to occur. The principal problems associated with thermal desorption in electron ionization are (1) involatility of large molecules, (2) thermal decomposition, and (3) excessive fragmentation.
The method, or mechanism, of electron ejection for positive ion formation proceeds as follows:
The sample is thermally vaporized.
Electrons ejected from a heated filament are accelerated through an electric field at 70 V to form a continuous electron beam.
The sample molecule is passed through the electron beam.
The electrons, containing 70 V of kinetic energy (70 electron volts [eV]), transfer some of their kinetic energy to the molecule. This transfer results in ionization (electron ejection) with the ion internally retaining usually no more than 6 eV excess energy.
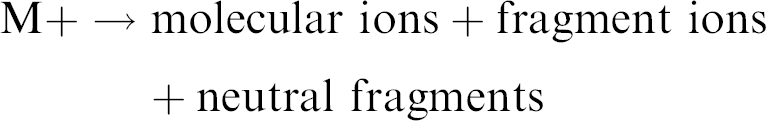
Excess internal energy (6 eV) in the molecule leads to some degree of fragmentation.
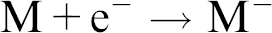
Electron capture is usually much less efficient than electron ejection, yet it is sometimes used in the following way for high sensitivity work with compounds having a high electron affinity:
Chemical Ionization (CI)
CI is applied to samples similar to those analyzed by EI and is primarily used to enhance the abundance of the molecular ion. Chemical ionization uses gas phase ion-molecule reactions within the vacuum of the mass spectrometer to produce ions from the sample molecule. The chemical ionization process is initiated with a reagent gas such as methane, isobutane, or ammonia, which is ionized by electron impact. A high gas pressure in the ionization source results in ion-molecule reactions between the reagent gas ions and reagent gas neutrals. Some of the products of the ion-molecule reactions can react with the analyte molecules to produce ions.
Negative ion chemical ionization (NICI) typically requires an analyte that contains electron-capturing moieties (e.g., fluorine atoms or nitrobenzyl groups). Such moieties significantly increase the sensitivity of NCI, in some cases 100 to 1000 times greater than that of EI. NICI is probably one of the most sensitive techniques and is used for a wide variety of small molecules with the caveat that the molecules are often chemically modified with an electron-capturing moiety prior to analysis.
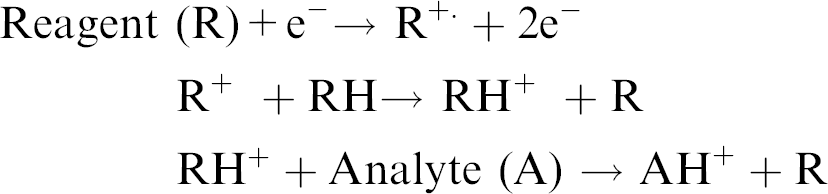
A possible mechanism for ionization in CI occurs as follows:
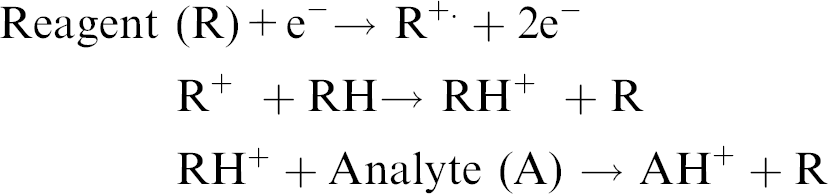
In contrast to EI, an analyte is more likely to provide a molecular ion with reduced fragmentation using CI. However, similar to EI, samples must be thermally stable since vaporization within the CI source occurs through heating.
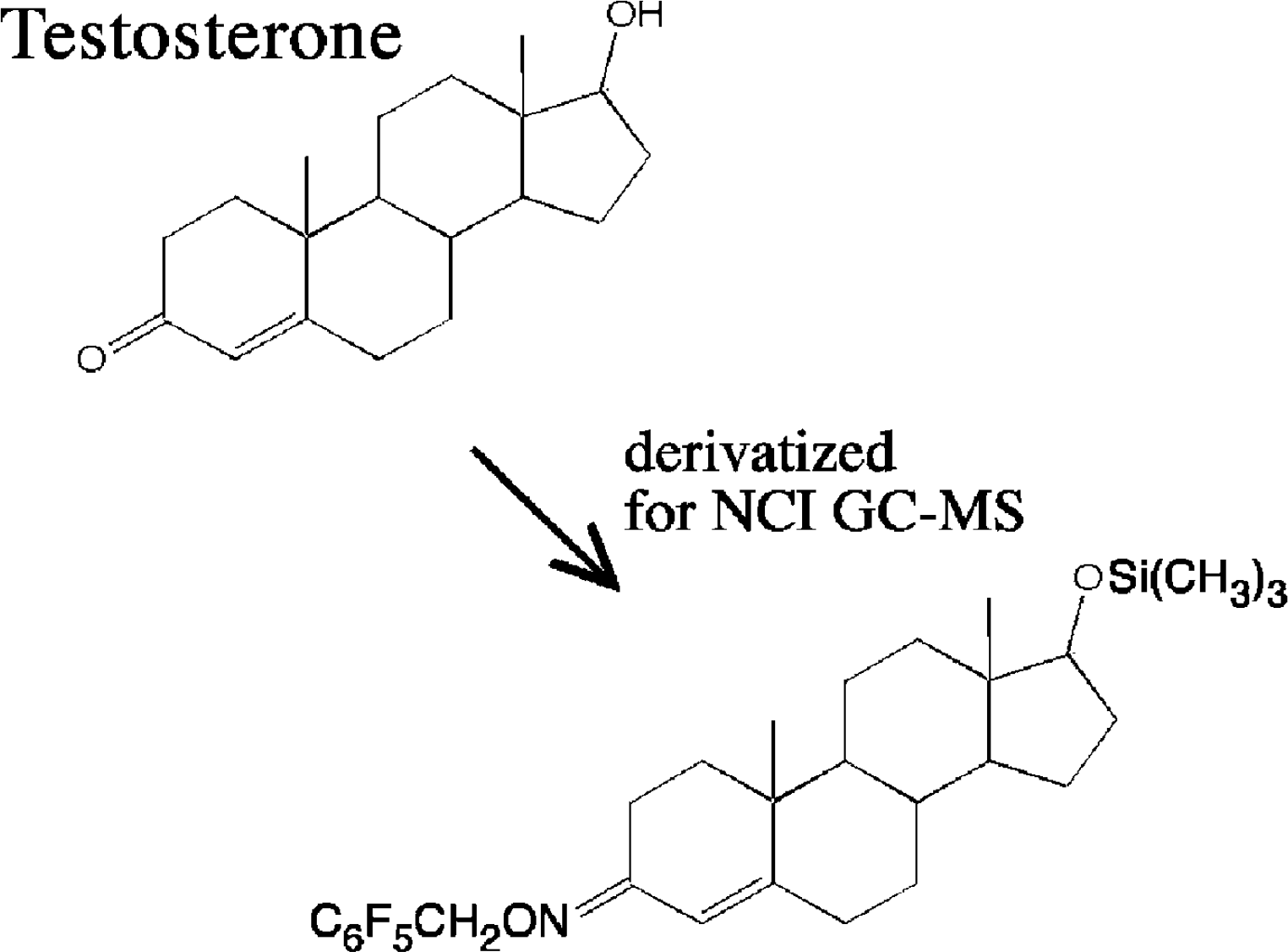
While most compounds will not produce negative ions using EI or CI, many important compounds can produce negative ions and, in some cases, negative EI or CI mass spectrometry is more sensitive and selective than positive ion analysis. In fact, compounds like steroids are modified (Fig. 19) to enhance negative chemical ionization (NCI).
The pentafluorobenzyloxime/trimethyl silyl ether derivatives of steroids make them more amenable to high sensitivity measurements using negative chemical ionization.
As mentioned, negative ions can be produced by electron capture, and in negative chemical ionization, a buffer gas (such as methane) can slow down the electrons in the electron beam allowing them to be captured by the analyte molecules. The buffer gas also stabilizes the excited anions and reduces fragmentation. Therefore, NCI is in actuality an electron capture process and not what would traditionally be defined as a “chemical ionization process.”
Overview
The mass spectrometer as a whole can be separated into distinct sections that include the sample inlet, ion source, mass analyzer, and detector. A sample is introduced into the mass spectrometer and is then ionized. The ion source produces ions either by electron ejection, electron capture, cationization, deprotonation, or the transfer of a charged molecule from the condensed to the gas phase. MALDI and ESI have had a profound effect on mass spectrometry because they generate charged intact biomolecules into the gas phase. In comparison to other ionization sources such as APCI, EI, FAB, and CI, the techniques of MALDI and ESI have greatly extended the analysis capabilities of mass spectrometry to a wide range of compounds with detection capabilities ranging from the picomole to the zeptomole level.