
Abstract
Primer walking of cloned DNA is a standard research tool. It has been used in the past to determine the sequence of individual clones of interest. With the expansion of DNA sequencing capacity the need to be able to walk larger numbers of clones has become necessary.
Our laboratory is a mid-sized genomics facility. In conjunction with the Advanced Biomedical Computing Center (ABCC) we have developed methods for automating the primer selection, DNA sequencing, contig assembly and sequence analysis for clones arrayed in microtiter format. This approach has allowed us to walk 475 clones (five microtiter plates) selected from a cDNA library.
Rationale
cDNA libraries frequently require sequencing. Due to their insert size (500–2000bp), they don't lend themselves to a typical shotgun sequence regimen employing universal primers. In order to complete a full-length clone sequence, it is desirable to begin in the vector and build contigs by designing new primers near the 3′ end of the read.
Some libraries can contain upwards of 20,000 clones. The sheer size of a project of this size can be daunting without an automated process for the orderly picking of insert specific primers, accumulation of individual clone data and the automated assembly of the contigs produced. Equally helpful is an automated ‘end of clone marker’, which can be defined by an algorithm binning all possible ‘clone finished’ statements.
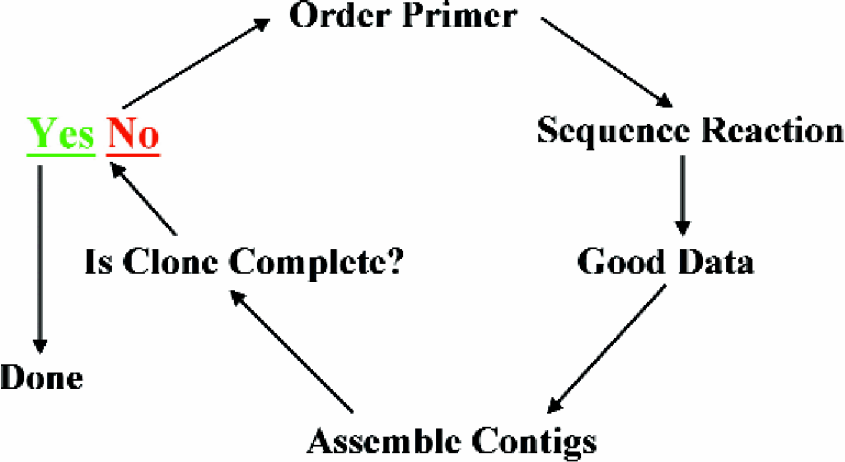
Process Overview.
AUTOMATION OF PRIMER WALK
To sequence short unknown DNA clones, begin with the vector primer and read into the insert.
Follow this by selecting the next primer that will continue the read and include overlap to permit accurate assembly.
Repeat as many times as necessary to complete the sequence. This is defined by reading back into vector and/or the poly(A) addition signal.
AUTOMATION OF PRIMER PICKING
Transfer all sequence reads from each plate run to single folder in LIMS system
Develop tool to batch auto pick primers in each folder based on optimal sequencing stringencies
Review auto-choice primer and accept/decline
Output tab delimited text for ordering according to primer vendor's specifications.
Request primers at constant concentration in deepwell plate
Review delivery to ensure proper positioning and orientation in plate.
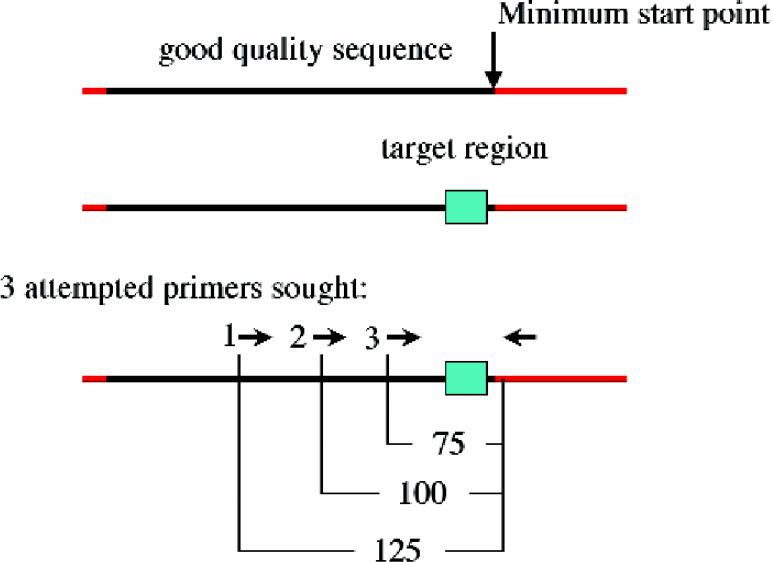
Primer Design Schematic.
PRIMER PICKING PIPELINE
Extract Chromats from LIMS folder (getChromats.pl)
Get base calls and quality values (phred)
Determine 3′ end of good quality regions (checker.pl)
Print primer input file (fldrPrimer.pl)
Run primer program on all sequences (primer 0,5)
Parse resulting output file into excel format (parsePrimer.pl)
Order primers and continue next cycle
Rerun primer with different parameters for unfound subset

An example of good quality read length, as shown by the q20 line and a polyA signal sequence followed by the actual poly(A).
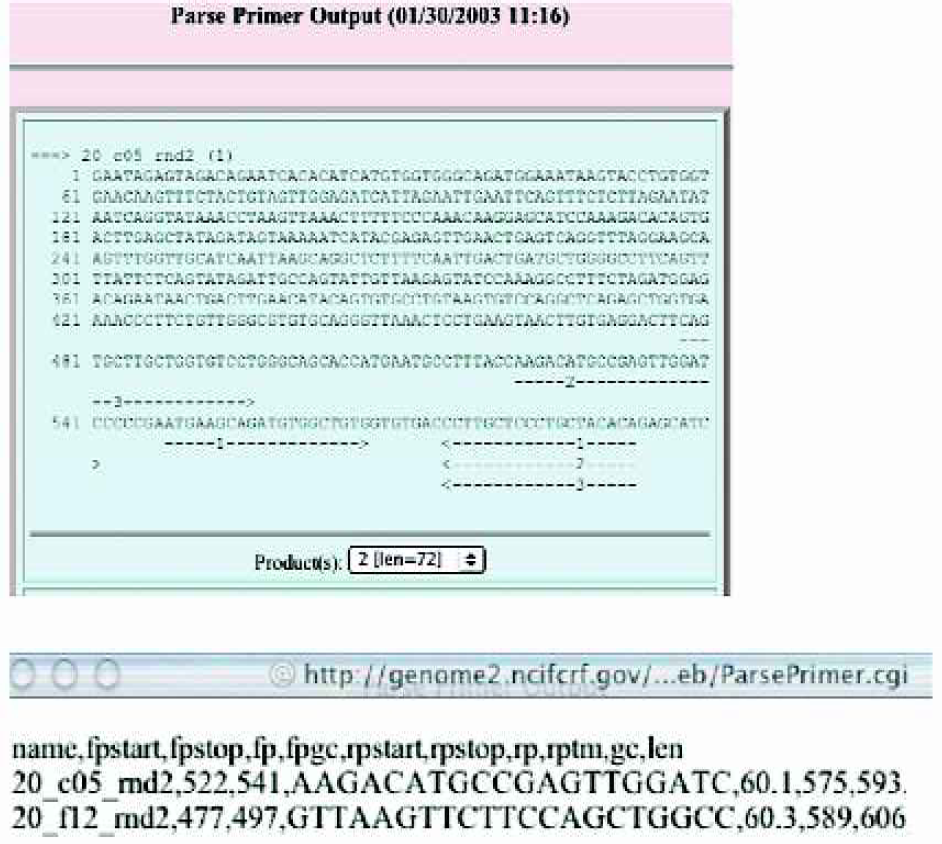
Primer picker parse output for choosing best of 3 possible primers and the final delimited output for ordering the next round.
SEQUENCE COMPLETION TAGS
One or more of the following criteria were used to define complete:
poly(A) addition signal (AATAAA) and poly(A)
Read through poly(A) into vector sequence
Read into long, >50+, poly(A)
SEQUENCE CONTINUATION TAGS
Long reads with high q20 values
Short reads with high q20
Poor quality reads - low q20
Repeat sequence and/or Order new primer more 5′ of previous primer
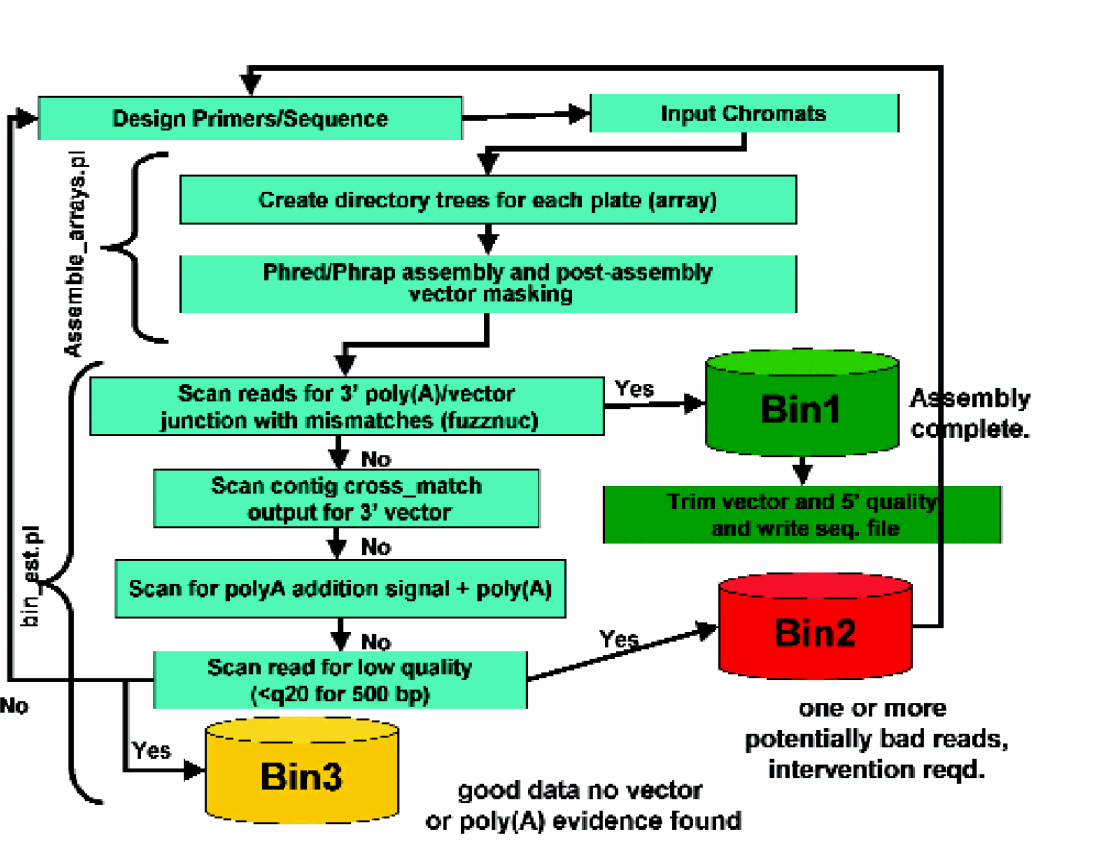
High throughput cDNA assembly/analysis pipeline.
Pitfalls of the Process
cDNAs - The poly(A) often denotes the end of sequence read but defining the end of real poly(A) and simply a long ‘A’ stretch in the clone is clouded due to the fact that the polymerase has trouble reading through most homo-polymeric regions. Also, as much as 30% of any given cDNA library will not show true polyA signal because the oligo-dT used to pull out the cDNA has actually laid down in an anomalous ‘A’ rich region. The library should be directionally cloned - but, that information may either be unavailable to you or incorrect. As a result, the first read of any library should be made on a single plate to determine the orientation.
Sequence - With a large number of cDNA, any library may not have the average insert size ‘advertised’. A mixture of shorter and longer inserts on a plate will cause the inefficient use of sequencer time by running partial plates.
Lack of an adequate LIMS to track the re-array of plates multiple times will adversely impact the progress of the job as well.
Conclusion
We have developed an efficient way to sequence large numbers of short clones that would not otherwise be good candidates for shotgun libraries. cDNA libraries are the ideal reagent to develop high throughput primer walking methods.
*This project has been funded with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. NO1-CO-12400.