Abstract
Advancements in the fields of genomic screening, molecular pathology and clinical research have resulted in a major increase in the demand for high quality DNA and RNA. This escalating demand has resulted in a sample preparation bottleneck and an emphasis on the development of new technologies to automate the purification process. Gentra has developed the AUTOPURE LS™ nucleic acid purification instrument, a platform capable of high-throughput sample purification from large samples, such as 10 mL whole blood. This article presents data showing the equivalency of DNA purified using manual and automated processing.
INTRODUCTION
Expanding on the groundwork laid by the human genome project, extensive studies are underway to document genetic diversity in human populations. 1,2,3 With the number of samples predicted to increase from millions to billions, there is a need to automate the process of purifying DNA and RNA from biological specimens. 4,5 In the fields of pharmacogenetics and clinical diagnostics, the most common source of DNA is whole blood. Because blood is collected typically in larger volumes, liquid phase purification methods, such as the PUREGENE™ DNA Purification Kit (Gentra Systems, Minneapolis, MN) are a common choice for molecular testing laboratories. Liquid phase chemistries have the advantage over solid phase chemistries in that they can be scaled linearly to accommodate both small and large sample volumes without sacrificing yield or purity. In addition, the high purity achieved with the well-established PUREGENE chemistry ensures that the DNA is stable and can be archived for future analyses, an important consideration for clinical trials and biorepository storage. This article presents the results of performance evaluations of the AUTOPURE LS, a robotic system which automates the PUREGENE DNA purification process for blood samples in the 1 to 10 mL volume range.
MATERIALS AND METHODS
Instrument Description
The AUTOPURE LS nucleic acid purification instrument automates Gentra's PUREGENE™ manual DNA prurification process and features on-board sample tracking, precision reagent dispensing, controlled mixing and pouring, gravimetric verification and centrifugation. Batches of 8 or 16 samples can be processed in two size ranges: 1–5 mL or 5.1–10 mL. Samples are poured into specially designed, 50 mL input tubes with bar coded red caps and placed into an input tube rack supplied with the system. An equal number of bar coded blue capped tubes are placed into the input rack to receive the purified DNA. After the operator places the input rack into the instrument and starts the purification process, the instrument begins by recording the input and output tube weights. Because sample volumes vary, the blood weights are adjusted with the first reagent, RBC Lysis Solution, to ensure that the centrifuge is balanced. The six main steps of the purification procedure are 1) lysis and removal of red blood cells 2) lysis of white blood cells to release the DNA 3) precipitation of proteins and other contaminants 4) precipitation of DNA in isopropanol 5) washing of DNA in 70% ethanol 6) re-hydration of DNA. The system runs Microsoft® Windows 2000 Professional software and outputs the data into Microsoft® Excel file format.
Sample Set Up
Blood samples were collected in 10 mL Vacutainer® Brand blood collection tubes (EDTA K3 No. 16852, Becton Dickinson, Franklin Lakes, NJ) and stored at 4°C until use. White cell counts were determined using a Coulter Counter® CBC-5 (Coulter Electronics, Inc. Hialeah, FL) calibrated using CBC-7 Hematology Controls (R&D Systems, Minneapolis, MN). DNA was purified from the blood samples within 96 hours of being drawn. DNA purified manually using the standard PUREGENE DNA Purification Kit (Cat. No. D-50K, Gentra Systems, Inc., Minneapolis, MN) was compared to DNA purified using the AUTOPURE LS instrument. DNA was purified from 16 whole blood samples using the AUTOPURE LS instrument according to the manufacturer's directions using PUREGENE reagents supplied (Gentra Systems, Inc., Minneapolis, MN). Upon completion of the purification process, approximately 80 minutes for 16 samples, the purified DNA was rehydrated overnight on a rotator (Clay Adams Nutator®, Fisher Scientific Catalog No. 14–062).
Genomic DNA Analysis
To assess DNA integrity, a volume of 2 mL was removed from each purified DNA sample and separated by 0.7% agarose gel electrophoresis. The DNA was electrophoresed for 1 hour at 75 volts using 0.125 μg/mL ethidium bromide in the gel and running buffer. The gel was photographed on a UV transilluminator using a Kodak Digital Imaging System EDAS 120 LE (Rochester, NY). Lambda DNA digested with Hind III was used as a size reference.
UV Absorbence Determination
DNA yields were determined using a Beckman DU-64 UV spectrophotometer (Fullerton, CA). A 10 μL volume of each DNA sample was diluted in 190 μL ultrapure deionized water and vortexed at high speed for 5 seconds. To obtain the DNA concentration, the 320 nm reading (background) was subtracted from the 260 nm reading, and the resulting value was multiplied by the DNA extinction coefficient of 50 μg/ml. Then, DNA yield was calculated by multiplying each concentration by the respective DNA volume as estimated by weight in grams. The A260/A280 ratio was determined by subtracting the background (A320) reading from each of the two readings before they were divided.
DNA Restriction Enzyme Digestion
DNA quality was evaluated further by analyzing digestion with a panel of 6 restriction endonucleases. In a digest volume of 25 μL, 2 units of EcoRI, Hae III, Msp I, Pst I, Hind III, Xba I (New England Biolabs, Beverly, MA and Sigma, St. Louis, MO) were added to 1 μg DNA and allowed to digest for 30 minutes at 37°C. To examine the samples for digestion, a volume of 2 μL was analyzed by 0.7% agarose gel electrophoresis as described above.
DNA Amplification
Purified DNA was evaluated for amplification performance using primers specific to a 1.5 kb target in one of the cytochrome P450 genes (CYP2D6 locus, 6 ). For each reaction, a quantity of 100 ng DNA was amplified in a 25 μL volume containing: 1X Taq Polymerase Buffer, 0.05 U/μL Taq Polymerase, 1.5 mM MgCl2, and 0.2 mM each dNTP (Promega, Madison, WI) as well as forward and reverse primers at 1 μM (Research Genetics, Huntsville, AL). The amplification conditions for the CYP2D6 target were: 30 cycles: 94°C 30 seconds, 63°C 30 seconds, and 72°C 30 seconds; 72°C hold 6 minutes, 4°C hold. A volume of 10 mL from each reaction was analyzed by 2% agarose gel electrophoresis as described above.
DNA Cross-contamination Analyses
DNA cross-contamination was examined by loading eight 10 mL blood samples alternately with eight 10 mL blank (phosphate buffered saline) samples and processing the 16 samples as a group. A 134 μL volume of DNA carrier (Glycogen Solution, 20 mg/mL, Gentra Systems, Inc.) was added to the output tubes to maximize precipitation of small quantities of DNA. To detect contaminating DNA in the negative control samples, a sensitive amplification assay was performed using primers specific for a 390 bp target within the HLA-H locus. 7 Each reaction was set up as described above and the samples amplified using the following conditions: 94°C for 5 minutes; 35 cycles of 94°C for 30 seconds, 58°C for 30 seconds, 72°C for 30 seconds; 72°C for 7 minutes, 4°C hold. Following amplification, 10 μL of each sample were loaded into a 2% gel and examined for the presence of contaminating DNA in the negative control samples. DNA standards were amplified concurrently to establish detection limits of the amplification assay; fewer than 10 copies of contaminating DNA are detectable with this assay.
RESULTS
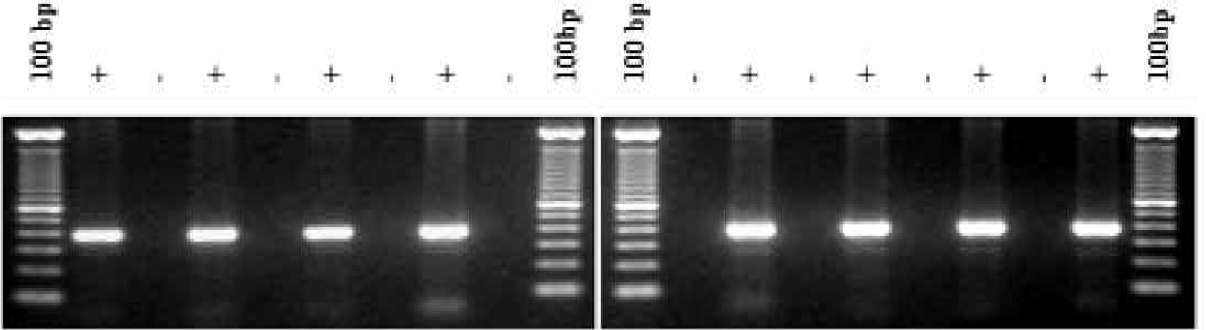
DNA samples purified using the standard manual PUREGENE method were compared to DNA samples purified on the AUTOPURE LS instrument (Figure 1) using replicate human whole blood samples collected from a single donor. Electrophoretic analysis showed that the DNA samples purified on the instrument were equivalent in molecular weight in comparison to the DNA samples purified manually (Figure 2). All samples appeared undegraded as shown by the abscence of low molecular weight DNA. The samples were also analyzed spectrophotometrically to determine DNA yield (Figure 3) and A260/A280 ratio (Figure 4). DNA yields were 348.3 (+/- 4.3) μg DNA (n=8) for the manually purified samples and 350.2 (+/- 5.1) μg DNA (n=8) for AUTOPURE LS purified samples. The A260/A280 ratios were essentially identical for DNA samples purified with either the manual or the automated method: 1.839 +/- 0.007 (n=8) for manual samples and 1.840 +/- 0.016 (n=8) for AUTOPURE LS samples. The DNA samples were also digested with six restriction enzymes and showed no difference in endonuclease digestion ability (Figure 5). Another measure of DNA quality was PCR amplification performance. Again, there was no detectable difference in amplification ability between the DNA samples purified manually and those purified on the AUTOPURE LS instrument for the 1.5 kb CYP2D6 target (Figure 6).

The AUTOPURE LS Automated Large Sample Nucleic Acid Purification Instrument.

DNA purified with manual PUREGENE and AUTOPURE LS is equivalent in size and quality. DNA was purified from replicate 10 ml human whole blood samples and processed in parallel using the manual PUREGENE method and the AUTOPURE LS instrument and subsequently analyzed by gel electrophoresis. The outside lanes contain λ x Hind III molecular weight markers. The remaining lanes contain replicate samples as indicated.
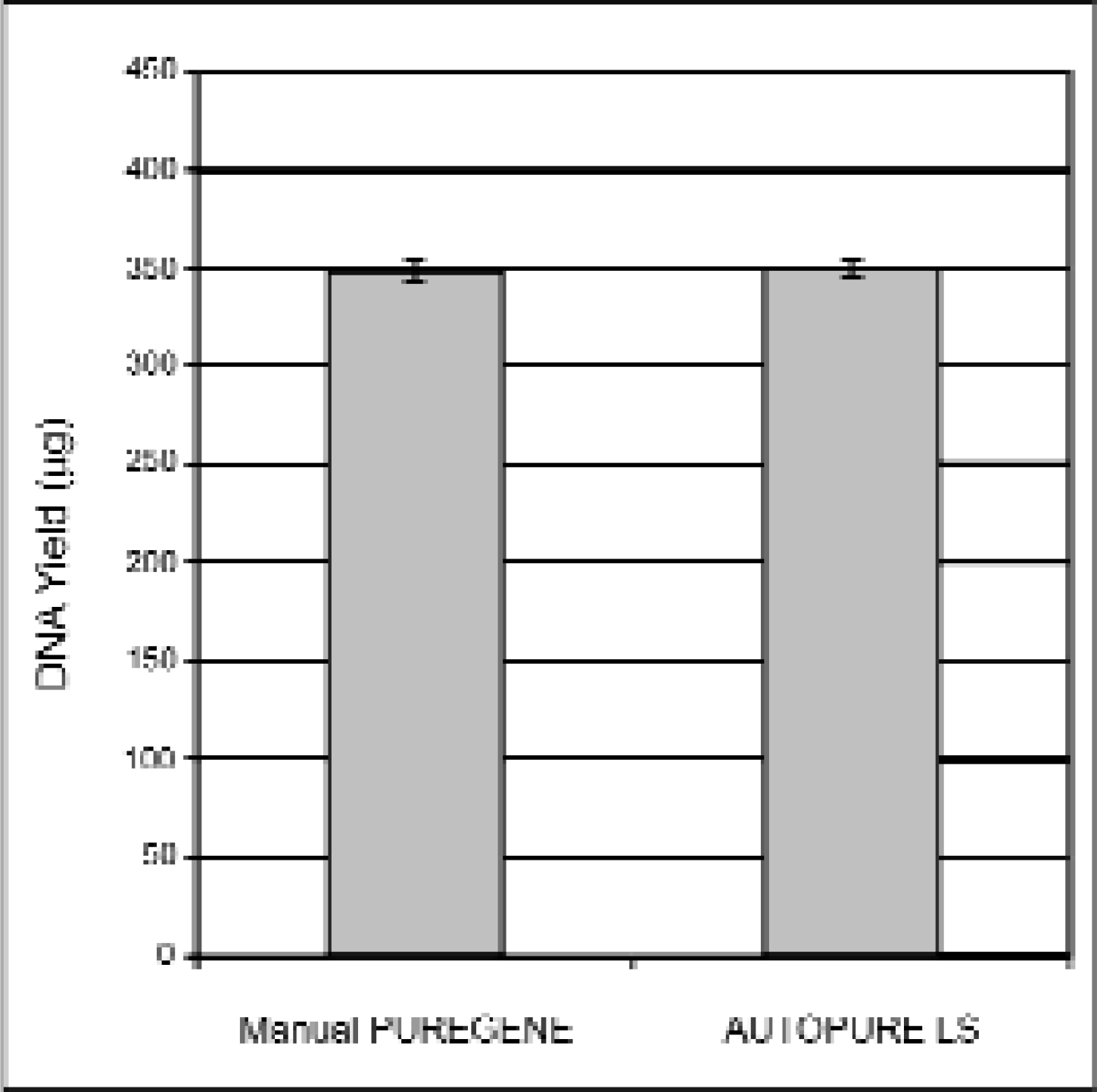
DNA purified with manual PUREGENE and AUTOPURE LS results in equivalent DNA yields. DNA was purified from replicate 10ml human whole blood samples and processed in parallel using the manual PUREGENE method and the AUTOPURE LS instrument. Purified DNA was then quantitated by UV spectrophotometry. DNA yields (+/-S.D.) are plotted in the graph above as indicated.
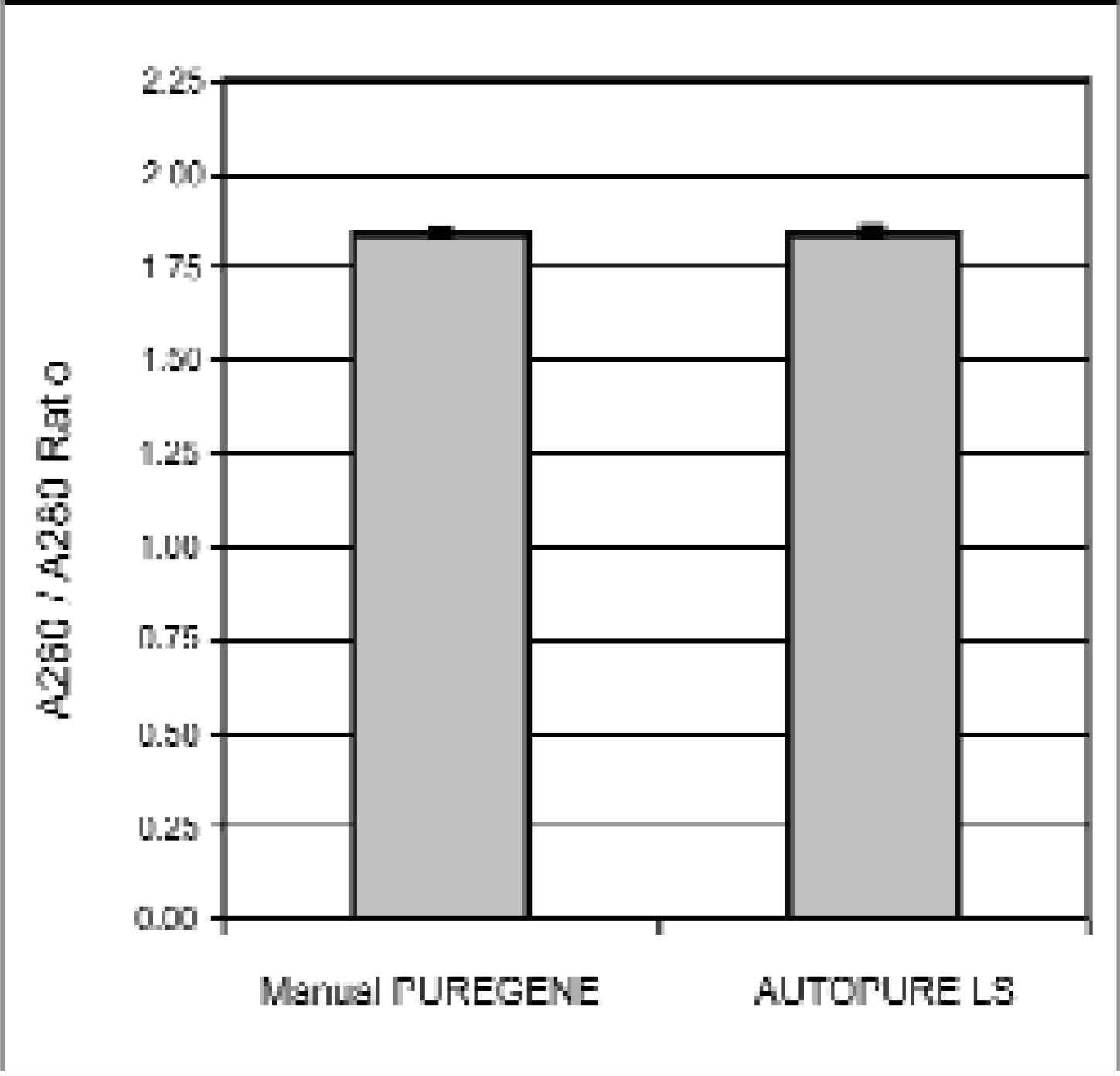
DNA purified with manual PUREGENE and AUTOPURE LS results in DNA with equivalent A260/A280 ratios. The A260/A280 ratios were determined by spectrophotometric analysis of the samples shown in Figure 2.

DNA purified with manual PUREGENE and AUTOPURE perform equivalently LS in restriction endonuclease digestions. DNA was purified from replicate 10 ml human whole blood samples and processed in parallel using the manual PUREGENE method and the AUTOPURE LS instrument. Purified DNA was then digested with restriction endonucleases as indicated.

DNA purified with manual PUREGENE and AUTOPURE LS perform equivalently in PCR amplifications. DNA was purified from replicate 10 ml human whole blood samples and processed in parallel using the manual PUREGENE method and the AUTOPURE LS instrument. A 1.5 kb amplicon from the CYP2D6 locus was amplified from the purified DNA.
Because blood samples are batch processed in groups of 8 or 16 on the AUTOPURE LS instrument, it was important to demonstrate that cross-contamination from adjacent samples does not occur. An experiment was set up to detect the presence of DNA contamination by processing blood samples adjacent to saline negative control samples. A sensitive HLA-H locus amplification assay was performed that detects less than 10 copies of contaminating DNA. The results, shown in Figure 7, demonstrate that no DNA was observed in the saline negative control samples, indicating the absence of detectable cross-contamination.
In conclusion, the performance of the AUTOPURE LS nucleic acid purification instrument was compared to the manual PUREGENE DNA Purification process and no significant differences in quantity, quality or performance of the purified DNA were detected. The AUTOPURE LS instrument should prove useful for those laboratories performing large sample nucleic acid purification where yield, quality and stability of the purified samples are critical.