
Abstract

SAMPLE ACQUISITION AND CONTROL
Biological sampling is important for drug discovery applications and for the basic biochemical and neurochemical discovery process. Several issues arise when sampling from biological environments. First, important biology can occur in small volumes (nL-pL) and at low concentrations (μM-nM). In order to access this information, the important biochemicals must be detectable and the time of specific biochemical events must be recorded. This means that a sampling and analysis method should be selective for the analyte(s) of interest, without loss or dilution, for efficient analysis. Tremendous accomplishments in biological sampling and analysis have been made, both for sampling and microfluidic manipulation. This article focuses on advances in controlling small volumes of biological samples and the analysis of specific analytes in those samples.
FLOW CONTROL BY RADIAL VOLTAGE
Fluid control of minute amounts of material is of paramount importance to the sampling and analysis of biological systems. Analysis of materials on a small scale has mostly been accomplished with the aid of electrokinetic effects, typically in a capillary electrophoresis (CE) setting. However, the electroosmotic flow (EOF) used in CE to manipulate fluids has proven difficult to control, therefore research has focused on improving electroosmotic flow control. To address the issues of flow control in CE two platforms are used: the microscale, or microfluidic chip approach; and the conventional, or capillary, approach.1,2 Because of the importance of EOF to this work, a brief overview of the flow mechanism is given here (also see Figure 1).
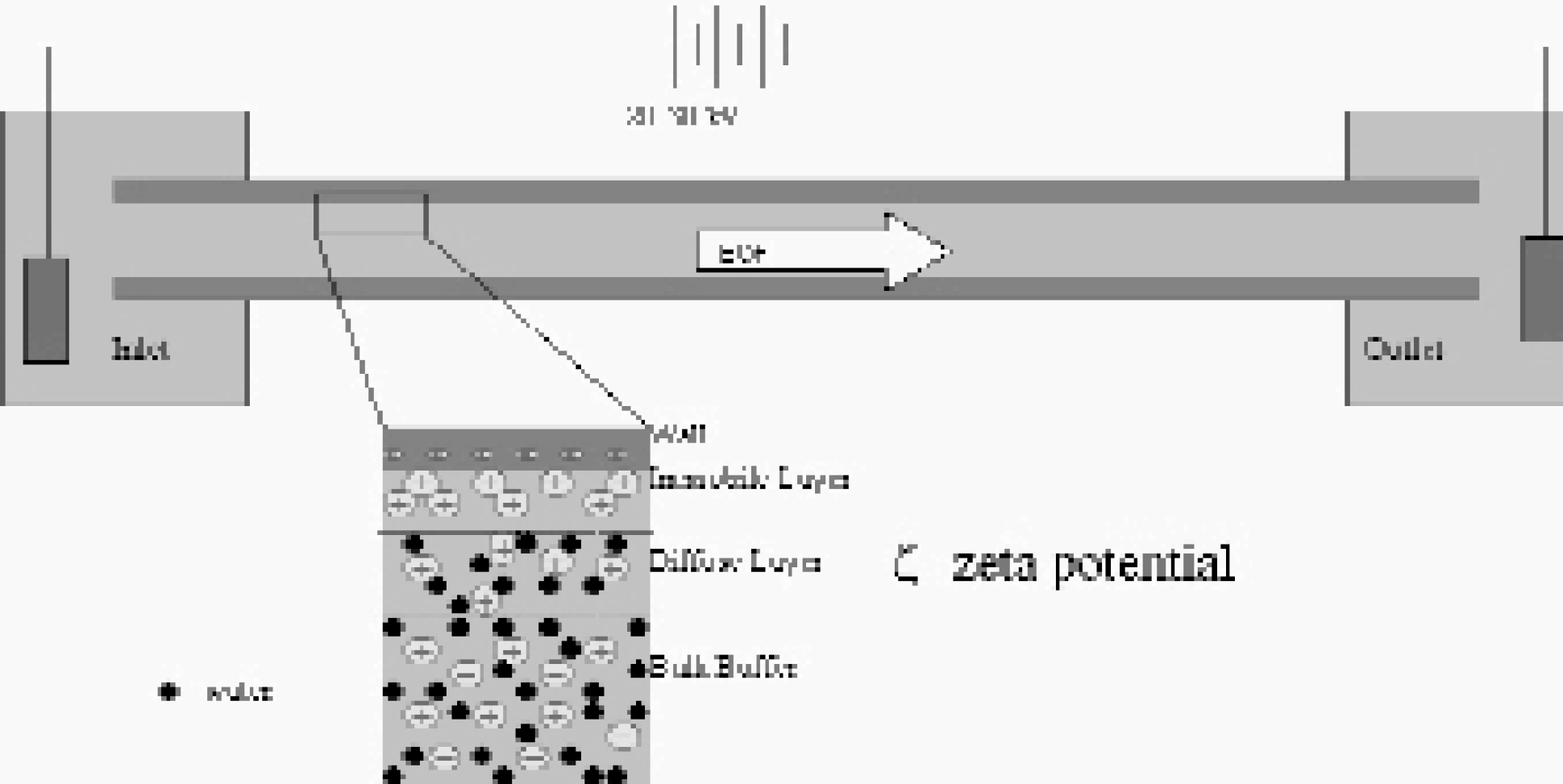
The formation of a charged layer at the capillary wall results in a potential drop (the z potential) near the wall, as well as EOF. The flow is generated when the cations near the wall move in the presence of an electric field applied to the capillary.
Electroosmosis is the flow mechanism generated when a buffer interacts with a charged solid interface, such as the capillary wall in CE, in the presence of a lateral potential field. 3 Above pH 3, the wall is negatively charged from the de-protonation of the surface siloxyl groups. The negative charge at the wall attracts cations from the solution. Some of the cations lose their hydration spheres and adhere to the surface (the immobile layer), while some remain hydrated and are free to move despite the attraction to the wall (the diffuse layer). The formation of this layer of charge results in a potential drop near the wall, termed the zeta (ζ) potential. In the presence of the lateral electric field, the diffuse layer will move toward the more negative electrode. It is this interplay between the wall, the solution, and the applied potential fields that creates EOF. 3
Control of EOF is essential to the resolution, efficiency, and usefulness of separations in both conventional and microscale systems. 3 For these systems to have true flow control, it is necessary to be able to change the flow rate in response to system conditions. Methods of flow control focus on altering the solution/wall interface by changing either the nature of the wall or the composition of the buffer. The use of coatings and buffer additives are common techniques to achieve these changes. 1 However, these methods are static and cannot be readily changed in situ. The use of a secondary voltage applied perpendicular to the separation axis, termed the radial voltage (RV), allows for dynamic change. 4 The application of this secondary voltage changes the charge on the wall. By changing the charge on the wall, the potential drop across the diffuse layer is altered and therefore the ζ-potential is changed. The consequence of changing the ζ-potential is a change in flow rate (υEO); 3
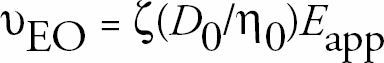
where D0 is the permitivity of the solution, η0 is the viscosity of the fluid, and Eapp is the applied axial electric field. Flow rates can be altered or even reversed using this method. 4
Studies in conventional capillary systems have shown large variability in the amount of flow control afforded by RV (Figure 2). 2 Investigations into the cause of this variability are ongoing, but it is probable that the source of the variation is the method by which the radial voltage is applied. In the microscale chip format, work by Schasfoort et al., has proven to be consistent with published conventional findings (changes in flow rate on the order of 1 × 10−5 cm2/V*s). 5 Work in our lab by Polson et al., however, is some forty-times more efficient than published systems (Figure 3). 1 Preliminary investigations into the source of this variability in the microchip format suggest that it is due to the porosity of silica after etching with hydrofluoric acid. It is suspected that this porosity optimizes the effects of the radial voltage.
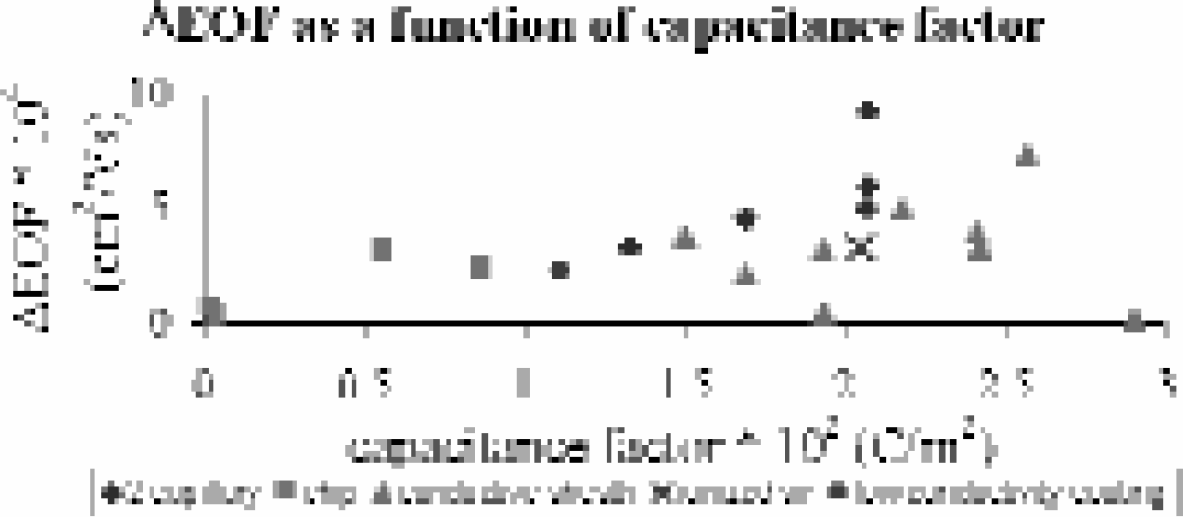
The variability in EOF control arising from application of a RV. Data is taken from published literature and identified here by the method of RV application.
WALL COATINGS
Another principle concern in flow control is the adsorption of molecules to the capillary wall. 6 When molecules adsorb to the capillary wall, the ζ-potential is changed and therefore EOF changes, making it difficult to reproduce results. Additionally the adsorption of sample molecules fundamentally changes the peak shape and concentration profile of the sample. Adsorption has been addressed primarily by using coatings on the capillary wall. 6 While coatings have been used for a multitude of other purposes, their function in relation to flow control for biological sampling is two-fold. First, the coating helps to prevent adsorption to the wall by masking the native charge, which helps to maintain a sharp peak profile and the native concentration of the sample plug. Second, the elimination of charge allows more efficient control of the flow by RV.3,7
Coatings previously developed for CE, based on the silicon-oxygen bond, have short lifetimes or limited pH range stability due to the susceptibility of silicon-oxygen bonds to acid and base catalyzed hydrolysis. 7 In order to overcome these limitations, a wall coating must be developed with improved stability in pH extremes as well as a long lifetime. Our current efforts in this area focus on titanium dioxide. Titanium dioxide has similar chemistry to silicon, which would allow the use of previously developed coatings on top of this layer. 8 However, titanium dioxide is not susceptible to acid and base catalyzed hydrolysis, and exhibits long lifetimes when bound to silica substrates. Use of titanium dioxide as a primer for other coatings would allow longer lifetimes and greater stability, providing another important tool for flow control.
FLOW MONITORING
With improvements in flow control comes the need to monitor flow. In addition, because the primary focus of our work is biological samples, our ideal flow monitoring system would be non-invasive in order to preserve the samples' native analytes and their concentration profiles.
Current methods of flow monitoring fall into several basic categories: chemical markers, changes in buffer components or concentration, measurement of ζ-potential, weighted effluents, and optical techniques. 9 However, these techniques either alter the separation system by introducing new chemicals, add complexity to detection schemes, or require complex valves.
A new technique has been developed where changes in refractive index are measured by laser induced interferometric backscatter to provide flow rate monitoring. 10 This technique changes the refractive index of the solution by introducing a small heat plug into the system. This heated plug is then detected a known distance away by the subsequent change in the backscatter pattern, and the flow rate is calculated using the distance and time of travel. We have shown this technique to be effective in capillaries for monitoring flow rates, and recent work by Swinney et al., has shown the detection mechanism's utility in microchip channels.9,11
SAMPLE PRECONCENTRATION
With the ability to control and monitor flow, one of the challenges that remains for biological sampling is the low concentration of most analytes in a specific biological environment. Previous methodologies to deal with this issue have included a variety of approaches, from using detection methods with improved sensitivity or increasing the optical path length for analysis by using larger capillaries to pre-concentration techniques such as sample stacking, field amplification, isotachophoresis, and chromatographic methods. 12
An alternative approach to improving sensitivity that we have investigated involves counterbalancing the flow rate in a capillary or microchannel against the electrophorectic mobility (EOM) of the analyte of interest. 12 By exactly matching the EOM of the analyte with an equal but opposite pressure flow, the analyte can concentrate at the capillary entrance (Figure 3A). Molecules can be collected at the entrance of a flowing stream, and subsequently released into the channel for detection when a sufficient concentration is obtained. This collection process is known as electrophorectic focusing. Electrophorectic focusing can be used online, requires neither alteration of the sample nor complex buffer systems, and selects a limited range of analytes (due to the specificity of the EOM) to be concentrated prior to detection. An example of electrophoretic focusing with carboxylate-modified beads is shown in Figure 3B.

Schematic showing the collection of particles at the entrance to the capillary during an electrophoretic focusing experiment. This result is achieved by balancing the bulk flow of the fluid against the electrophoretic mobility of the analyte.

Fluorescent micrography of 200 nm carboxy-late modified beads collecting at a capillary (20 mm i. d./150 mm o. d.) under the influence of induced vacuum (0.040 atm). The image on the left shows no electrophoretic migration of the beads; the one on the right shows migration and collection of the beads after 4 minutes (-14 kV applied at the capillary tip)
APPLICATION TO BIOLOGICAL SAMPLES
With a better control of flow, the ability to monitor it, and the ability to preconcentrate analytes of interest if necessary, we have investigated several biological systems and analytes that are of interest to the pharmaceutical and clinical world. In addition, the potential of these techniques to be miniaturized allows the possibility of mass production and automated analysis. Below we detail several ongoing studies that are contributing valuable insights into the problems and providing solutions for analyzing biological samples.
PROCESS CONTROL MEASURES FOR LIPOSOMES
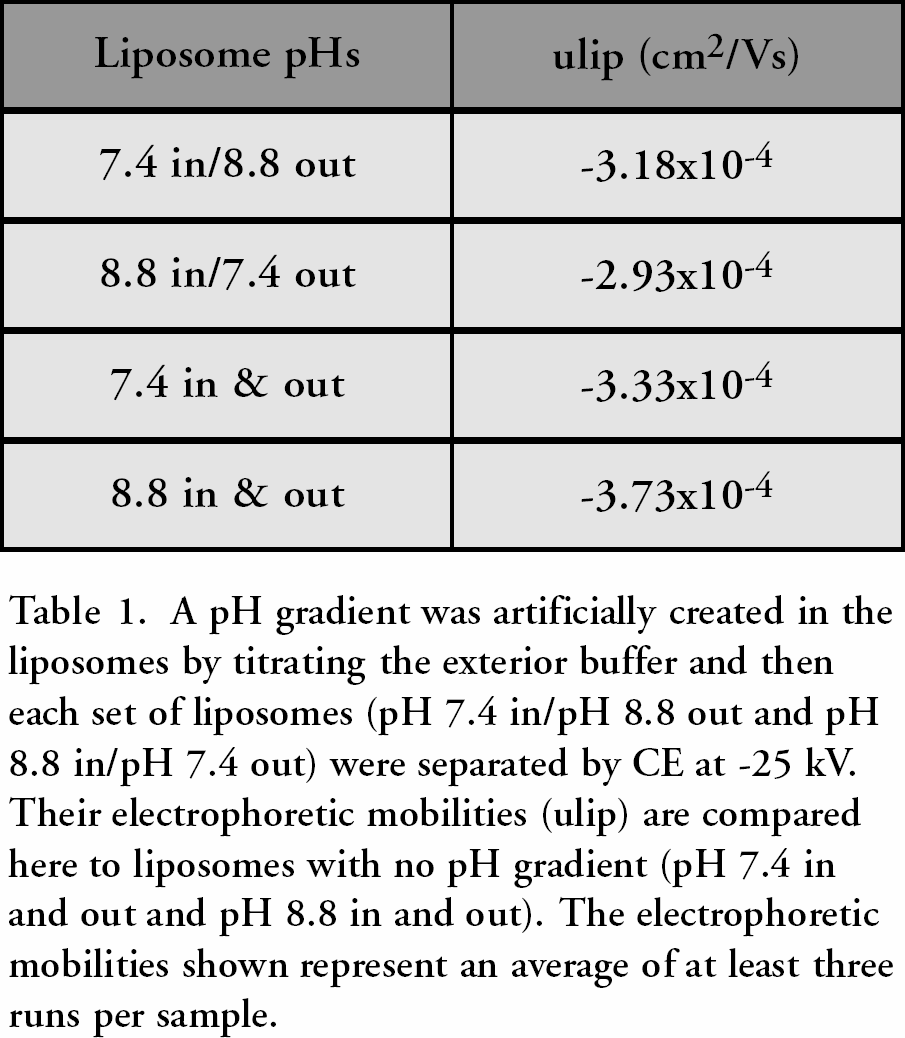
Liposomes are interesting because of their widespread use in the pharmaceutical and cosmetics industries, their ease and variability of formulation, and their stability. 13 Many questions about their properties and how those change under external stimuli or with composition remain unanswered, therefore we have undertaken a study of their properties by CE. Specifically, liposomes used in artificial photosynthesis are being studied to determine how their surface properties and electrophoretic mobilities change when a pH gradient is created across the membrane. 14 Since the interior aqueous cavity is electrically neutral, it is expected that it should have no effect on the exterior surface charge. However, our results suggest that if a different concentration of protons exists in the interior aqueous cavity as opposed to the exterior bulk buffer, the surface charge on the liposome changes. This is suggested by the difference in electrophoretic mobilities observed for liposomes with different pH gradients across them (Table 1), as the electrophoretic mobility of the liposome is directly proportional to its surface charge. Current work is focusing on quantitatively correlating these changes in electrophoretic mobility to the pH gradient existing across the membrane.
Another interesting experiment was suggested by this work. If the interior aqueous cavity did affect the surface charge, it was possible that the charge was being transferred from the interior to the exterior of the liposomes. Liposomes with and without a pH gradient across their membranes were run separately by CE, then mixed together and run again. Instead of seeing two separate peaks as expected for two separate populations in the mixed sample, only one peak was observed with an electrophoretic mobility that was not equal to either population's electrophoretic mobility alone. This suggests that the two populations are distributing the pH gradient present in one population initially across both of the liposome populations. Ongoing studies are attempting to determine if this is in fact the case; and if so, by what mechanism this equilibration occurs.
A pH gradient was artificially created in the liposomes by titrating the exterior buffer and then each set of liposomes (pH 7.4 in/pH 8.8 out and pH 8.8 in/pH 7.4 out) were separated by CE at −25 kV. Their electrophoretic mobilities (ulip) are compared here to liposomes with no pH gradient (pH 7.4 in and out and pH 8.8 in and out). The electrophoretic mobilities shown represent an average of at least three runs per sample
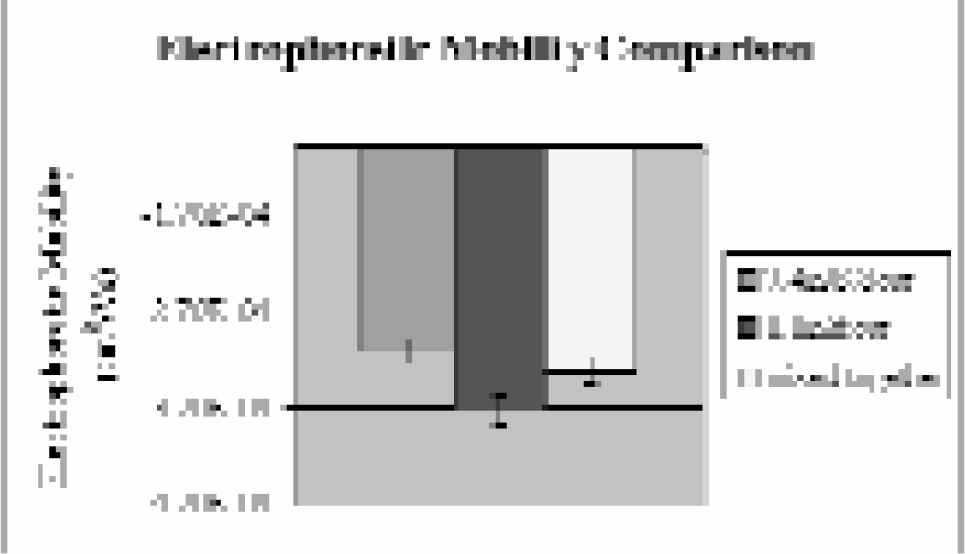
A comparison of electrophoretic mobilities of two liposome samples, pH 7.4 in/pH 8.8 out and pH 8.8 in and out, when run alone (purple and burgundy, respectively) and when mixed together (yellow). Note the mixture of the two samples results in an electrophoretic mobility that is not equal to either's electrophoretic mobility alone.
Clearly, more studies need to be done to ascertain the behavior of liposomes. It is important to note, however, that our results do support our belief that CE provides a quick and quantitative means of monitoring the properties and behavior of liposomes, and thus may be exploited for process control measures.
MINIATURIZING THE CLINICAL ASSAY
To improve on the analysis times and reduce the sample volumes required in clinical assays, we have chosen to miniaturize the immunoassay, which has the advantage of highly specific analyte recognition and can provide quantitative kinetic data. Our approach is a flow-based immunoassay that is adaptable to microchip and capillary formats already in use: a paramagnetic (PM) bead-based immunoassay. PM beads have previously been used as a solid support phase in sandwich-type immunoassays, but this is the first reported flow-based microimmunoassay utilizing the paramagnetic beads.15,16 In this case, a magnet is used to hold the PM beads in place as they are flowed through a conventional capillary or microchip channel, thus no specialized chip fabrication is required. The beads are labeled through a biotin-avidin linkage with a primary (capture) antibody, and once a bed has been packed, the sample containing the analyte of interest (the antigen) is flowed through the bed for a designated period of time. Detection is then accomplished with a fluorescein-labeled secondary antibody.
This assay has several advantages. First, it minimizes the required sample size and reagent quantities, thus reducing cost. Reaction and analysis times are up to 40 times faster than conventional immunoassays, and the design and technology are readily adaptable to automation. It is also amenable to any chip or capillary-based system already in use. It requires no specialized reagents, as antibodies are commercially available for many biological analytes of interest and can be biotinylated or tagged with any desired fluorophore for the researcher's purposes. Lastly, the microchip can be used again for conventional CE analysis or a different PM bead-based immunoassay by removing the magnet and washing the paramagnetic beads away; or can be fabricated in a cheap plastic for disposability.
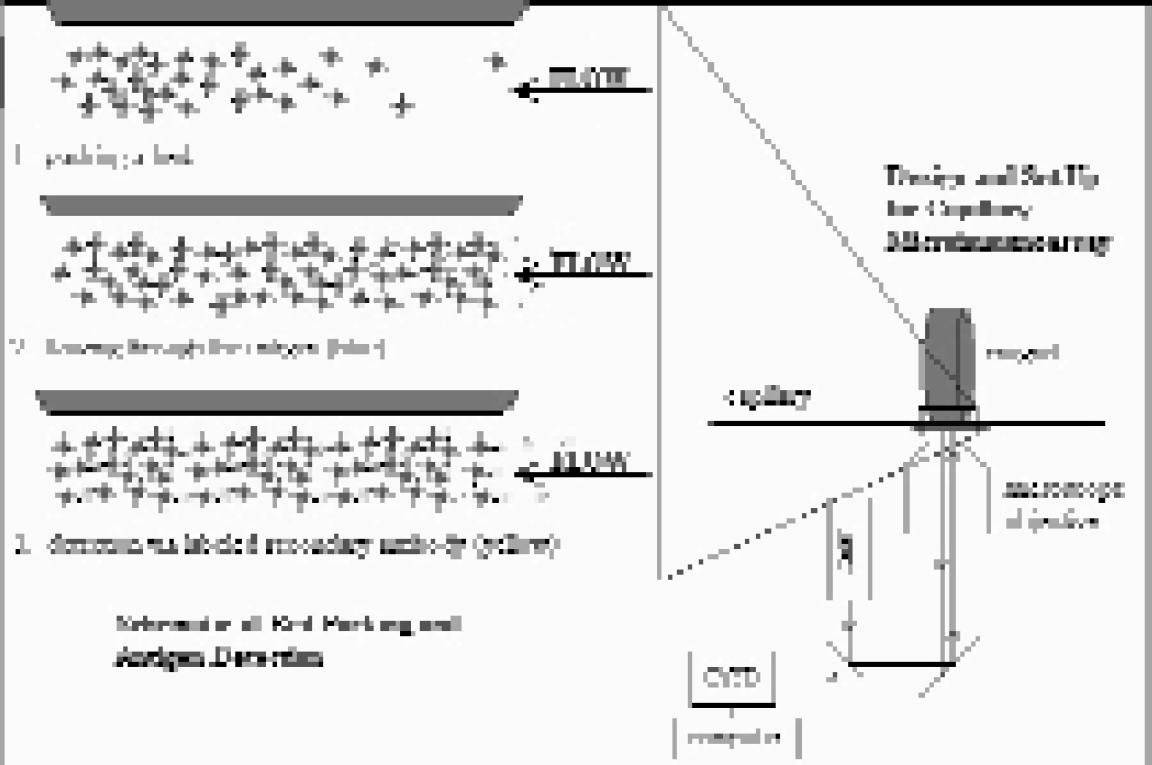
Set up and basics of bed packing and antigen detection. Complementary antibody pairs are used for antigen capture and detection. Since complementary antibodies are already used in traditional immunoassay formats such as ELISA, specialized antibody generation and preparation is not necessary.
The viability of this PM bead-based approach has been demonstrated with three different systems. Initial studies were performed with a pair of anti-fluorescein (FITC) antibodies, where the FITC itself was the antigen. These studies demonstrated that 90% of the maximum signal was achieved after only 3 minutes of exposure to FITC (Figure 6). However, the detection limit of 10μM for FITC suggested possible problems with detecting biological analytes, which are typically at much lower concentrations (nM to pM range). This relatively high detection limit was attributed to quenching of the FITC from the antibody, as the FITC itself was interacting with the antibodies, and has been previously reported in the literature.(17)
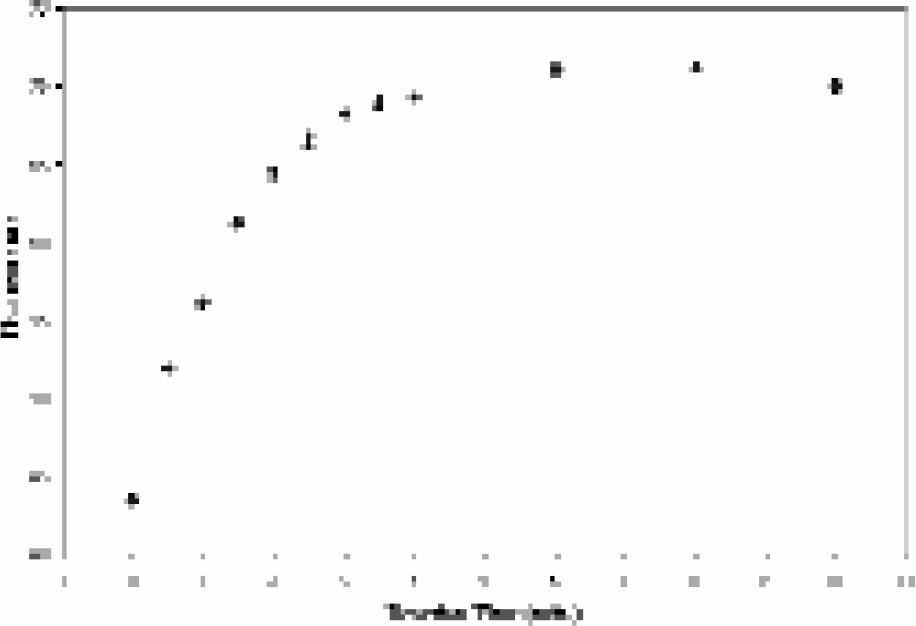
Residual fluorescence signal obtained from reaction of FITC with anti-FITC antibody four minutes into buffer wash.
To test this hypothesis, a biological analyte of interest was selected and examined as a test case using a sandwich type assay within the PM bead-based system. Parathyroid hormone (PTH) was chosen, as clinicians commonly assay for its presence in patients exhibiting symptoms of a thyroid disorder. Data shown in Figure 7 demonstrate the ability to assay PTH at a concentration of 160 pM, which is at the high end of biological concentrations. Recent work has focused on another biological molecule of interest: interleukin-5, a known asthma and inflammatory response marker. Preliminary results have demonstrated the viability of the technique (Figure 8), and we are currently making improvements to the detection scheme to provide assays at clinically relevant concentrations in a fraction of the time taken with conventional microtiter plate-based immunoassays.
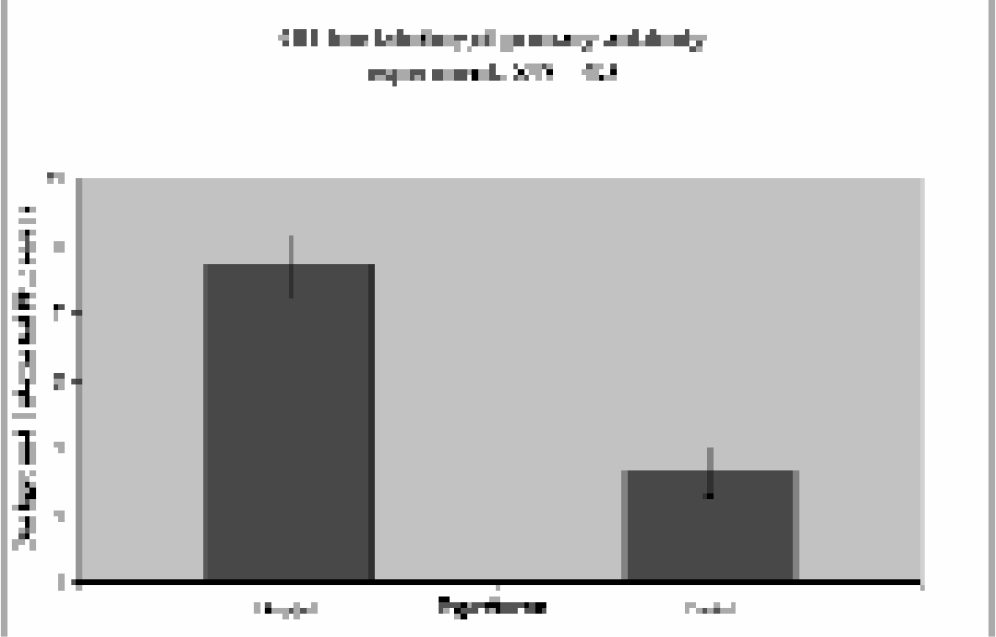
Signal obtained from sandwich microimmunoassay of parathyroid hormone (PTH) at a concentration of 1.4 ng/mL. Data was taken four minutes after a buffer wash was started. In this case the incubation of the PTH with the antibody-labeled paramagnetic beads was done off capillary, but similar results are obtained for on line incubation.
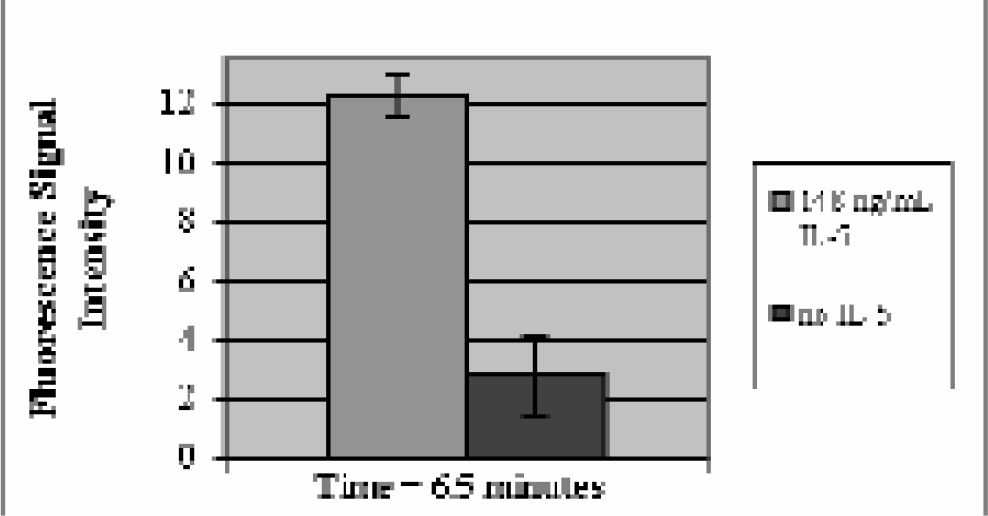
Preliminary result for on line incubation of interleukin-5 (IL-5) using the flow-based microimmunoassay. Data was taken 6.5 minutes after a buffer wash was initiated. The biotinylated and fluorescein labeled antibody pair was purchased commercially and used as received, demonstrating the ease of reagent preparation and commercial availability.
DIRECT SAMPLING FROM BIOLOGICAL TISSUES
Ultimately clinicians and other medical personnel would like a virtually non-invasive sampling process whereby molecular markers of certain conditions found in certain tissues or cell types can be assayed with minimal discomfort to the patient. Our vision of direct sampling would involve a capillary with a diameter less than or equal to a human hair being inserted into the tissue of interest and a picoliter volume sample of cells being extracted for analysis.
To this end another focus of our work is the direct sampling of biological tissues. Preliminary studies are being conducted with a model invertebrate system, Clione limacina. This work focuses on the separation and identification of previously unknown peptide hormones from the pleural ganglia that are thought to be involved in sexual maturity (Figure 9). This is accomplished by the direct sampling of cellular fluids through a 20 μm inner diameter (i.d.) fused silica capillary. Cellular fluids are mixed in the capillary with fluorescent tags known to react with the primary amines of peptides, then separated by CE and detected. Early data show that direct sampling of the desired ganglia can be accomplished and unknown peptides fluorescently tagged and separated (Figure 10). Future work in this area will concentrate on the isolation and identification of the peptides. Once this technique has been improved, further studies will include direct sampling for interleukin-5 from awake, freely-moving mice.

Picture of direct sampling of right pleural ganglion of Clione limacina with a 23 mm external diameter capillary (black arrow). Also shown is the fluorescent sac of neurons located in the pedal ganglia which serves as a landmark for sampling the desired area (white arrow).
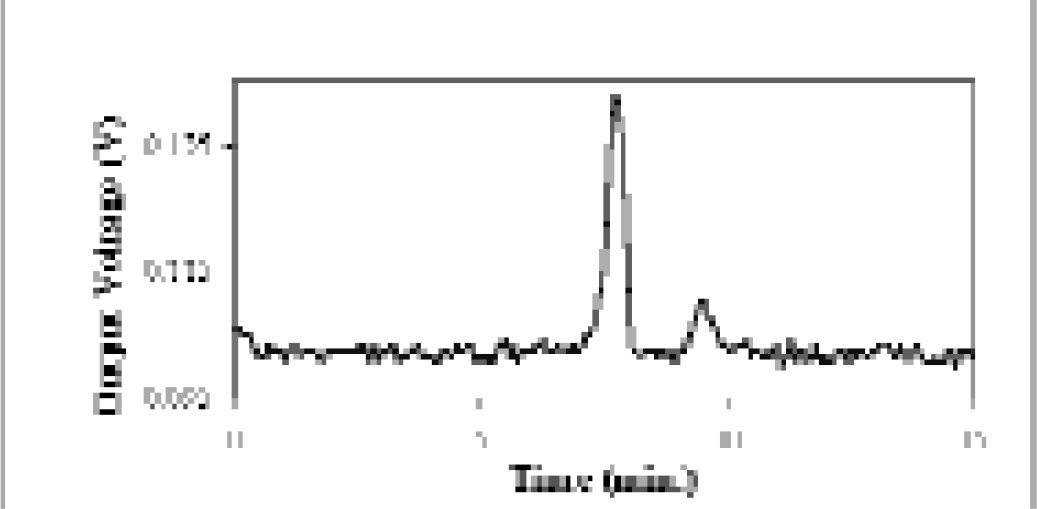
An electropherogram from the direct sampling of the right pleural ganglion of Clione limacina. Two unknown peptides were successfully labeled with a fluorescent tag in the capillary and separated by CE.
CONCLUSIONS
By combining our microchip-based immunoassays, flow control and monitoring, CE, and direct sampling, we aim to create novel microchips that are capable of process control and clinical analyses for a reasonable cost. With the current outstanding pace of developments across the industry and in academic laboratories, we have no doubt that analytical microdevices will play a very important role in laboratory automation.