
Abstract
DCM is classified among the most hazardous atmospheric pollutants and therefore, the development of safe methods for the disposal of residual DCM is of great importance. Activated carbon- supported catalysts containing platinum nanoparticles as the active phase, with a high proportion of metal in the zero-valent state, were prepared in order to obtain catalysts of high performance in the gas-phase hydrodechlorination (HDC) of dichloromethane (DCM). Catalysts with Pt particles of sizes as low as 1.6 nm per average metal particle were obtained, leading to a high effectiveness in the dechlorination of effluent. All catalysts showed high initial dechlorination activity; however, a lower surface acidity in the support led to a higher stability of the conversion during the operation, as it avoids coke formation. When increasing metal load up to 2% (w/w), platinum nanoparticles of similar sizes originated, while a greater amount of Pt0 was obtained; this process therefore improved dechlorination activity to a significant extent. DCM conversion up to 90% and selectivity for non-chlorinated products higher than 90% were achieved. The influence of DCM concentration in the feed and the H2/DCM molar ratio was also investigated.
1. Introduction
Chloromethanes (CMs) are volatile organic compounds (VOCs) widely used in the chemical and pharmaceutical industries as solvents and reactants, as well as in aerosols and adhesives and extraction processes, among others. Nevertheless, they are considered pollutants due to their high stability (remaining in the environment for several years) and toxicity [1-3], and therefore, their emission is subjected to strict legal regulations. Growing attention is being paid to air pollution originated by waste treatment, with a bigger focus on sustainable development and green management [4-6]. The emission of CMs into the atmosphere contributes to the destruction of the ozone layer, to the formation of photochemical smog and to global warming. Dichloromethane (DCM) is classified among the most hazardous gas pollutants, because of its high toxicity and carcinogenic character. Hydrodechlorination (HDC) constitutes an emerging technology for the treatment and recycling of chlorocompounds [7]. It operates at moderate conditions, leading to much less harmful reaction products, which can be recovered or destroyed by incineration. Precious metals are active catalysts for carbon-chlorine bond cleavage [8, 9]. The use of catalysts based on different precious metals and acting as supports for the HDC of chloromethanes has been reported. Palladium, platinum, rhodium and ruthenium have been tested as active phases, while SiO2, Al2O3, TiO2 and carbon materials have been tested as supports [10-19]. Palladium catalysts were the most active catalysts, with DCM conversions up to 100% and high selectivity toward non-chlorinated compounds (65–100%); however, they suffered severe and rapid deactivation. Pd/Al2O3 catalysts have been investigated by several authors [12-14] and have shown low activity in the hydrodechlorination of dichloromethane (<30% atmospheric pressure, even at 300°C); however, selectivity toward non-chlorinated compounds of up to 100% have been obtained. Other authors [10, 11, 18] have studied this reaction in the context of several catalysts and using different metals and supports, obtaining the best results with Pd and Pt catalysts, with conversions near 100% and complete dehalogenation. However, in these studies, whether the effect of time on stream was not investigated, or the catalysts were rapidly deactivated. Aristizabal et al. [15] and Gonzalez et al. [16, 17] reported high activity for Pd/TiO2 catalysts; however, these were strongly deactivated after 48 hours on stream. Pt is commonly reported to be less active than Pd, with conversion values lower than 80% and somewhat less selective to non-chlorinated compounds, but shows better stability [18-21]. On the other hand, rhodium and ruthenium have been less studied. These catalysts showed lower activity than those of Pd and Pt (Ordonez et al. [10] reported DCM conversion lower than 60%), or alternatively, suffer deactivation [22] in the same manner as palladium catalysts. In previous studies [1, 22-24], we investigated the HDC of DCM using catalysts based on Pd, Pt, Rh and Ru on activated carbon. Although all the metals noted above were shown to be effective in the HDC of the chloromethanes, significant differences were found in both activity and selectivity. Among them, platinum supported on activated carbon (Pt/C) is considered extremely useful for the hydrodechlorination of dichloromethane, because of its high selectivity to non-chlorinated compounds and its superior stability.
Catalytic activity, selectivity and stability are related to catalyst structural properties and many variables can affect to these characteristics, such as preparation procedures and the supports used. The oxidation state of metallic particles on the catalyst's surface and metal particle size play important roles in the catalytic activity of these catalysts in the context of hydrodechlorination [1, 7, 22-27]. The properties of the active phase depend not only on the metal precursor, but also on the type of support used.
The catalyst support provides a surface on which to disperse the metal, allowing for higher metal surface areas, which can favour catalytic activity and likely, also selectivity and stability. The effect of the support on the hydrodechlorination of organochlorinated compounds has been analysed from different perspectives such as metal-support interaction, support porous structure and acid-base properties. In a recent review [7], the literature related to supported metal catalysts for hydrodechlorination reactions was investigated. This study shows how metal particle size, support acid-base properties and electronic interactions with the supported metal can contribute to catalytic response.
Both inorganic and organic materials can be selected as supports. Among them, TiO2, MgO, Al2O3, SiO2, modified zeolites [28-32] and activated carbon are most frequently used in hydrodechlorination [18, 33-35]. However, inorganic materials can be attacked by the HCl formed in the HDC reaction, leading to an increase of surface acidity, which exacerbates the effects of HCl poisoning and coke deposition. Activated carbons are appropriate supports for this reaction, since they are relatively inert to the HCl formed, although the performance of activated carbon is dependent on different variables, such as pore-size distribution and surface oxygen groups.
As stated above, promising results were found for the gas-phase hydrodechlorination of DCM with Pt/C catalysts, as a high stable Pt/C catalyst was found [22, 23]. However, the dechlorination degree of the effluent can be increased by the optimization of the catalyst and the reaction conditions. It was found that the formation of small metal particle sizes and high proportions of metal in the cero-valent state were key factors for the stability of the catalysts, because they prevent the oligomerization of chloride radicals with subsequent coke formation, chloromethanes adsorption and HCl poisoning. Nevertheless, in previous studies only a type of carbon was used as support. Therefore, the influence of other properties like the surface acidity of carbon in the stability of the catalyst and the role of carbon surface chemistry and its interaction with Pt particles in the formation of zero-valent Pt nanoparticles was not investigated, which are of great significance for the design of effective and stable catalysts.
For these reasons, the aim of this study was to prepare carbon supported catalysts containing Pt nanoparticles with high proportions of Pt0, but modifying the nature of the carbon surface and Pt loading in order to analyse the influence of these parameters – especially carbon surface acidity – on the physico-chemical properties of the catalysts and their hydrodechlorination activity, selectivity and stability. Moreover, in order to extend the practical application of the technique, the performance of the catalysts was investigated in a wide range of DCM concentrations and H2/DCM ratios in the feed.
2. Experimental Procedures
Carbon-supported platinum catalysts containing 1% w/w Pt nominal load were prepared by incipient wetness impregnation of three commercial activated carbons (Erkimia S.A. (CE), Merck (CM) and Chemviron (CC)). In addition, four catalysts containing 0.5, 1, 1.5 and 2.0% w/w metal loading were prepared by the same method using Erkimia carbon as the support. A carbon particle size in the range of 0.25–0.50 mm was used. An aqueous solution containing a pre-calculated amount of metal salt (H2PtCl6 6H2O, supplied by Sigma-Aldrich) was adjusted to obtain the desired metal load and was prepared using a volume according to the retention volume of the corresponding carbon support. For example, a catalyst containing 1% w/w Pt load was impregnated with an aqueous solution containing 0.0265 g of salt/g of the catalyst. The retention volume of the supports was experimentally measured by adding water dropwise to the dried supports until the pores were filled, evidenced by the deposition of a thin water film in the external surface of catalyst particles. The retention volumes of the three carbons were 2.4 cm3 g−1 to CE, 2.0 cm3 g−1 to CM and 1.5 cm3g−1 to CC. The impregnation of the support was carried out by adding the solution of metal salt dropwise to the dried activated carbon while stirring. The samples were dried overnight at room temperature and heated to 100°C at a heating rate of 20°C h−1, the final temperature being maintained for 2 h. The catalysts were reduced in situ before use at 300°C under H2 flow (50 cm3 min−1) for 2 h. A heating rate of 10°C min−1 was used. Hydrogen was supplied by Praxair with a minimum purity of 99.999%.
The porous structure of the catalysts was characterized by N2 adsorption-desorption at −196°C (Tristar II 3020, Micromeritics). The bulk metal content was determined via inductively coupled plasma-mass spectroscopy (ICP-MS) in a Perkin-Elmer model Elan 6000 Sciex system, which was equipped with an autosampler (Perkin-Elmer model AS-91). Metal dispersion of the reduced and used catalysts was determined by CO chemisorption at room temperature (PulseChemiSorb 2705, Micromeritics). Transmission electron microscopy (TEM) of the samples was carried out using a JEOL JEM-2100F microscope operating at 200 kV. The surface composition of the catalysts was analysed by X-ray photoelectron spectroscopy (XPS) (5700C Multitechnique System, Physical Electronics), using MgKα radiation (hm = 1253.6 eV).
The activity of the catalysts in the hydrodechlorination of dichloromethane was tested in a continuous flow reaction system described elsewhere [1], consisting essentially of a 9.5 mm i.d. fixed bed micro-reactor, coupled to a gas chromatograph with a FID detector for the analysis of the reaction products. The experiments were performed at atmospheric pressure using a total flow rate of 100 Ncm3 min−1, a H2/dichloromethane molar ratio of 25–200, spacetimes (τ) of 0.8 and 1.7 kg h mol−1 and temperatures of 150°C-250°C. A DCM concentration in the feed of 1000 ppmv was used, except when the influence of this parameter was investigated. In such a case, the feed concentration varied in the range 1000–3500 ppmv. Blank experiments were performed at the same conditions than those of the experiments carried out with catalysts. A higher temperature of 250°°C and DCM concentrations of 1000 and 3000 ppm were used. The results showed that DCM conversion was insignificant in non-catalytic hydrodechlorination.
The catalysts' behaviour was evaluated in terms of dichloromethane conversion (XDCM) and overall dechlorination, turnover frequency (TOF) and selectivity toward the different reaction products (Si). Conversion is defined as the moles of DCM transformed per mol% in the feed and dechlorination as the mol% of chlorine removed from carbon compounds. Turnover frequency corresponds to the molecules of DCM transformed per atom of metal exposed and time. Selectivity to reaction products is defined as the moles of DCM transformed to give a particular product from the total moles of DCM reacted. The evolution of catalytic activity in terms of time on stream was examined in the long-term (26 days) experiments.
3. Results and Discussion
3.1 Characterization results
Table 1 summarizes the values of bulk metal content, BET surface area, micropore volume and platinum dispersion (percentage of surface Pt) of the fresh and used (26 days on stream in HDC of DCM) 1% Pt catalysts prepared with the different activated carbon supports.
Metal content, BET surface area and metal dispersion of the fresh and used Pt/C catalysts
All fresh catalysts showed a high BET surface area, although this was lower in the case of Pt/CM (Merck carbon). No significant differences were observed following use in DCM HDC, which discards the blockage of the porous structure by carbonaceous deposits as a cause of the deactivation of the catalyst, as will be explained in a later section of the text.
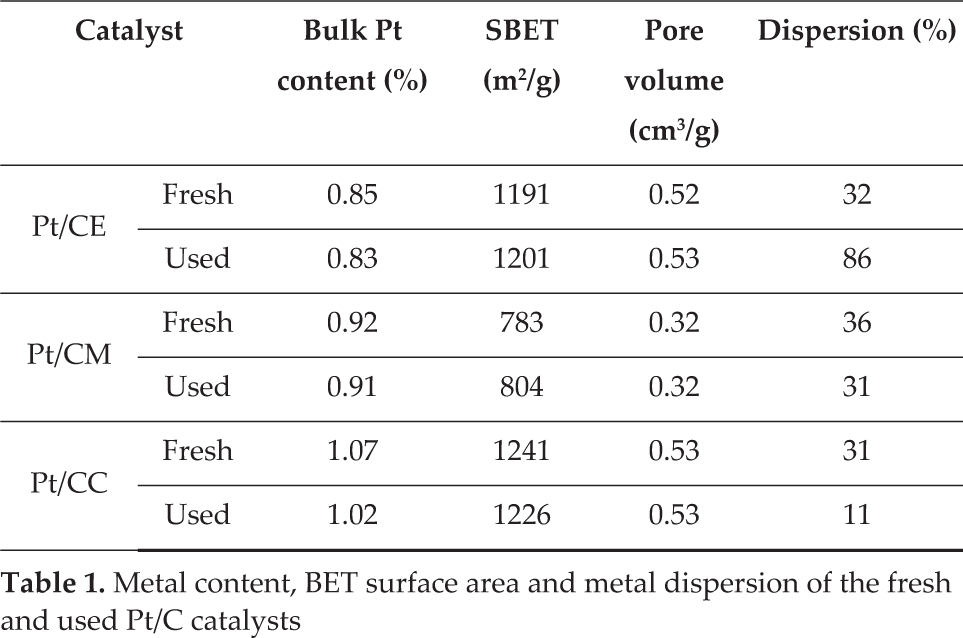
CO chemisorption results indicate that platinum was fairly well dispersed in all fresh catalysts, with slight differences among them. The high Pt dispersion can be attributed to the high surface area of the activated carbons and their surface chemistry, which provokes interaction between the active phase and support during catalyst preparation [1]. TEM images (Figure 1) and metal particle size distribution (Figure 2) confirmed the formation of Pt nanoparticles in the fresh Pt/CM and Pt/CC catalysts, with average size values of 1.6 and 2.0 nm, respectively. The formation of Pt nanoparticles was previously observed for a Pt/CE catalyst, though a slightly higher average value of 5.8 nm was obtained [23].

TEM image of the fresh-reduced (a) and used (b) Pt/CM catalyst and fresh-reduced (c) and used (d) Pt/CC catalyst
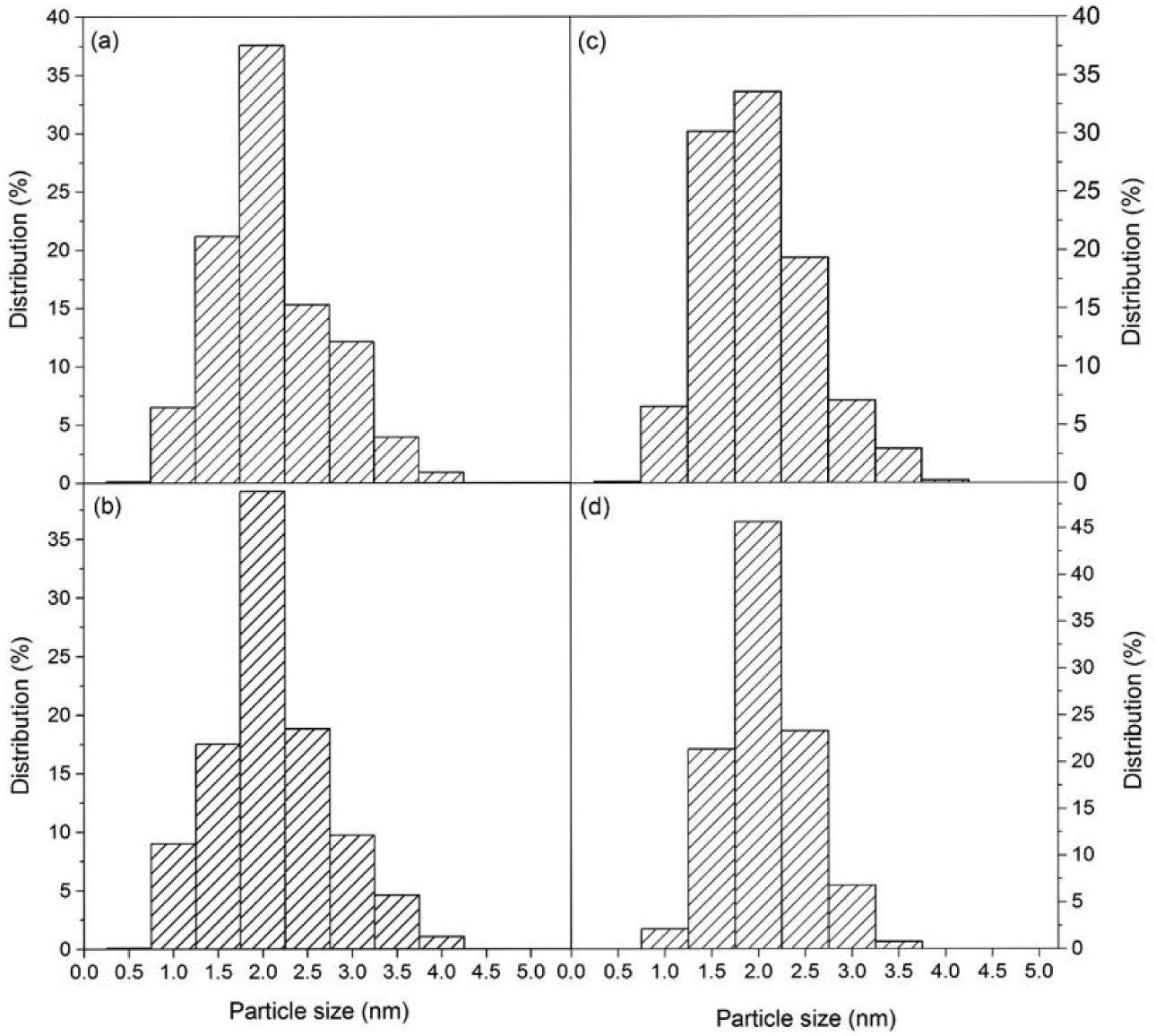
Platinum particle size of fresh-reduced (a) and used (b) Pt/CM catalyst and fresh-reduced (c) and used (d) Pt/CC catalyst
However, significant differences were found in the evolution of the dispersion during the hydrodechlorination reaction. The metal dispersion of Pt/CM essentially remained constant during the 26 days of the experiment, whereas substantial changes were observed in the cases of Pt/CE and Pt/CC (Table 1). The latter suffered an important decrease in platinum dispersion, which was consistent with the loss of activity showed by this catalyst and which will be discussed below. Contrarily, an important increase in dispersion was observed in Pt/CE, which did not show deactivation. This was attributed to the re-dispersion of platinum particles during the HDC reaction [23]. Several authors ascribed the re-dispersion of metallic particles to the effect of HCl, which they reported to provoke the formation and volatilization of unstable metallic chlorides, followed by metal re-deposition [28, 36]. Platinum will be reduced due to the high concentration of H2 introduced as reactant. TEM analysis of the used catalysts showed that no significant variations in the particle size and distribution were observed after 26 days on stream in the case of Pt/CM and Pt/CC. These results suggest that the drop in Pt dispersion in Pt/CC following DCM HDC must have been due to partial blockage of the active sites by (chloro) carbon species. Re-dispersion of Pt particles was also observed by TEM for Pt/CE catalysts [23], in agreement with CO chemisorption results.
Erkimia carbon (CE) was examined by N2-TPD to observe on the surface oxygen groups and the results were compared with those for published Merck (CM) and Chemviron (CC) carbons [37]. The amounts of evolved CO and CO2 and the CO/CO2 molar ratio are presented in Table 2. Acidic groups evolve as CO2[37]. The overall amount of desorbed CO2 and the CO/CO2 molar ratio provides information on the surface acidity of the active carbons. As can be seen in Table 2, the results obtained indicate that the surface of CC has a more acidic character than that of CE and CM, in this order. Deconvolution of TPD profiles supports this trend (Table 3). In the literature [38], it is generally accepted that a higher surface acidity favours higher metal dispersion, since the increase of acidic sites increases the hydrophilic character of the support. However, the differences observed in this case did not lead to significant variations of Pt dispersion. On the other hand, the acid-base properties of the support appeared to have a potential effect on catalyst stability. For example, Early et al. [39] reported that the more acidic the support, the more intense the deactivation of the catalyst in the hydrodechlorination of CFCs with palladium supported on Al2O3, fluorinated Al2O3 and AlF3. The deactivation appears to be caused by the formation of carbonaceous deposits on the surface of the catalyst, which is favoured by the acidic character of the support. Similar results were obtained in this contribution and will be explained in more detail later.
Results from TPD profiles for total evolved CO and CO2

Assessment of surface oxygen groups for the carbon supports from TPD

The Pt 4f region of the XPS spectra of all the catalysts showed a doublet corresponding to Pt 4f5/2 and Pt 4f7/2. Figure 3 depicts the deconvoluted spectrum of the Pt/CE, Pt/CM and Pt/CC catalysts when reduced and after DCM hydrodechlorination. Separation between the Pt 4f5/2 and Pt 4f7/2 peaks, due to spin orbital splitting, is a quantized value of 3.33 eV. The Pt 4f7/2 peak, lying at around 72.0 eV, can be attributed to Pt0 (zero-valent Pt), while the Pt 4f7/2 peak, located at around 73.4 eV, is related to Ptn+ (electro-deficient Pt) [22].

Deconvoluted Pt 4f spectra of Pt/CE, Pt/CM and Pt/CC catalysts before and after DCM hydrodechlorination
Table 4 shows the surface Pt composition (as determined by XPS) and the relative proportions of the Pt species of the fresh and used Pt/C catalysts. The surface concentration of chlorine and the relative amount of inorganic and organic chlorine obtained from the deconvolution of the Cl2p region are also included. The surface Pt percentages were quite similar in all the catalysts, Pt0 was always the prevailing species as a result of the reduction step included in the preparation procedure. The highest Pt0/Ptn+ ratio was found for Pt/CM, followed by Pt/CC and Pt/CE. This ratio diminished somewhat in the used Pt/CE and Pt/CM catalysts and the decrease was more pronounced in the case of Pt/CC, which suffered important deactivation during time on stream. These results confirm Pt0 as the main active phase for DCM adsorption [23]. Additionally, an increase in the relative amount of organic chlorine was found for this catalyst after HDC of DCM. This relationship will be discussed in more detail below.
Surface composition of the fresh and used catalyst from deconvoluted XPS profiles

Table 5 shows the bulk metal content, determined via ICP-MS, the Pt dispersion values, determined by CO chemisorption and the Pt0/Ptn+ ratio obtained by XPS (using the same procedure as described above) for the catalysts with different Pt load. The bulk metal content values were in good agreement with the nominal Pt load. Within the range tested, no significant differences in platinum dispersion were found when the platinum load was varied. All catalysts were fairly well dispersed, with values between 31% and 42%. However, the highest Pt0/Ptn+ ratio was found in the 2% Pt catalyst, whereas no variations were observed bellow this load (Table 5).
Bulk Pt content, Pt dispersion and relative distribution of Pt species on the surface of the catalysts with different Pt loads

3.2 Hydrodechlorination activity
3.2.1 Effect of carbon support
Figure 4 shows the DCM conversion values and global dechlorination achieved with Pt/CE, Pt/CC and Pt/CM catalysts at 1.7 kgcat h mol−1 space-time and different reaction temperatures. The error of the experiments was checked, having been lower than 5% in all cases. As can be seen, all the catalysts were extremely active for the detoxification of the effluent, with hydrodechlorination activity up to 80%. An important increase in DCM conversion with temperature was observed within the range tested (200°C-250°C). This increase was more pronounced in the cases of Pt/CM and Pt/CC, yielding Pt/CC the highest initial conversion (around 87%).

Initial conversion (symbols) and dechlorination (columns) of DCM (a) and yield to methane and monochloromethane (b) with Pt/CE, Pt/CM and Pt/CC
Hydrodechlorination of dichloromethane yielded with all the catalysts only methane and monochloromethane, with high selectivity toward methane (>80%) in all cases. The selectivity patterns can be related to the formation of nanoparticles in Pt. The small sizes of Pt particles avoided the formation of hydrocarbons higher in number of carbons than methane, bigger than methane, which were previously found to be stabilized in big metal particles [23, 24, 26] which may lead to stronger deactivation of the catalysts. Increasing the reaction temperature favours the formation of methane to the detriment of monochloromethane. Yield to methane increased significantly with reaction temperature, as shown in Figure 4.
As stated in previous research [23], the Pt/CE catalyst showed high stability during time on stream. This catalyst did not suffer appreciable loss of activity during 26 days of operation. In the same working conditions (250°C and 1.7 kgcat h mol−1), long-term experiments were performed with the Pt/CM and Pt/CC catalysts, the results of which are shown in Figure 5.

Overall dechlorination during time on stream with Pt/CE, Pt/CM and Pt/CC
Like Pt/CE, Pt/CM was active and highly stable in the hydrodechlorination of DCM, only a slight increase in DCM conversion was observed after 26 days on stream (a conversion value around 70% was attained). However, the Pt/CC catalyst suffered important deactivation during time on stream, decreasing DCM conversion from 90% to 50%. Selectivity toward methane and monochloromethane (the only reaction products) remained almost constant in all cases.
The high stability of Pt/CE was attributed to the small metal particle size following re-dispersion of Pt particles during the reaction and the high Pt0/Ptn+ ratio, which allows for a greater resistance to poisoning by chloromethane adsorption [23]. As indicated previously, Pt/CM and Pt/CC showed a very small and similar metal particle size prior to and after being used in the HDC reaction (Figures 1 and 2). Moreover, when the oxidation state of metal is compared, all of the reduced catalysts showed high Pt0/Ptn+ ratios and even Pt/CC showed a higher proportion of zero-valent Pt than Pt/CE, while the latter was indicated as being extremely stable. These results can explain the high stability of Pt/CM catalysts but not the deactivation of Pt/CC.
Pt/CC exhibited a significant decreased of Pt dispersion (from 31% to 11%) along the reaction, which provokes a loss of activity that cannot be attributed to the metal sintering of Pt particles, as this did not occur (see Figures 1 and 2). Nevertheless, in the case of Pt/CC, a significant decrease in the proportion of Pt0 was observed following the reaction by means of XPS (Table 4). This specie, Pt0, is the main active specie in the HDC of chloromethanes containing Pt/C catalysts, according to previous works [22, 23]. In addition, the Pt/CC catalyst showed a significant increase in the amount of organic chlorine following reaction, whereas no corresponding changes were found for the two other catalysts (Table 4). The increase in the proportion of organic chlorine appeared to be related to the poisoning of active sites with organochlorinated compounds [23-25]. The lower resistance to deactivation on the part of Pt/CC can be ascribed to its higher surface acidity, as evidenced by the N2-TPD experiments (Table 3). As indicated previously, the acidity of the support favours coke deposition and consequently, the deactivation of the catalysts, as it has been found in other hydrodechlorination reactions. Carbonaceous deposits can be formed rapidly on the more acidic supports and block active sites, leading to the loss of activity along time on stream [39].
3.2.2 Effect of metal load
Figure 6 shows the effect of Pt loading on the catalytic activity at different reaction temperatures (150°C-250°C) and 0.8 kgcat h mol−1 space-time. DCM conversion highly increased alongside the Pt content of the catalyst, primarily at the highest reaction temperatures, reaching conversion values around 70% at 250°C for the 2% Pt/CE catalyst. A more pronounced change was also observed, increasing the Pt load from 1.5% to 2.0%. Increasing the platinum content of the catalyst increased the number of available active sites, since the surface activity of carbon supports are capable of stabilizing Pt nanoparticles, even at higher Pt contents, therefor platinum dispersion was fairly similar in all cases (Table 5).

Effect of Pt content of Pt/CE on DCM conversion and selectivity to methane and monochloromethane at different HDC temperatures
The dechlorination of the effluent also increased alongside Pt content, due to the increase of methane selectivity. At 250°C with the 2% Pt/CE catalyst, more than 90% methane selectivity was achieved (Figure 6).
As shown in Table 5, an important increase in the proportion of zero-valent species was observed when increasing Pt load from 1.5% to 2.0%. However, no significant changes were observed below 1.5%. These results are consistent with the more pronounced increase of DCM conversion and dechlorination within the 1.5%-2.0% Pt range, since a higher Pt0/Ptn+ ratio favours HDC of DCM, as indicated previously, as well as methane selectivity.
3.2.3 Effect of feed concentration
Figure 7 shows the evolution of DCM conversion when increasing the DCM feed concentration at a space time of 0.8 kgcat h mol−1 and using the Pt/CE catalyst. As can be seen, the catalysts remain extremely effective and even a slight increase in DCM conversion was obtained when increasing DCM concentration.
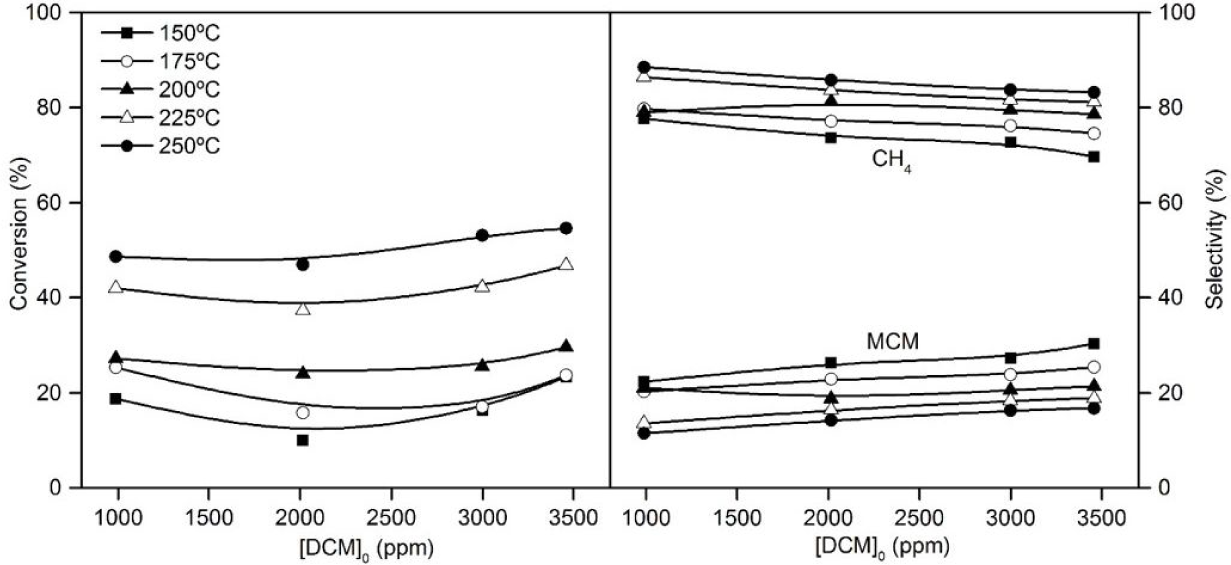
Effect of feed concentration on DCM conversion and selectivity to reaction products at different HDC temperatures
A similar product distribution was also observed when increasing the concentration of DCM in the feed and therefore, similar dechlorination activity of the effluent.
3.2.4 Effect of H2/DCM molar ratio
Figure 8 shows the evolution of DCM conversion when modifying the H2/DCM molar ratio at a space time of 0.8 kgcat h mol−1 and using the Pt/CE catalyst. As can be seen, the catalysts effectively used the H2/DCM molar ratios from 50; however, a remarkable increase in DCM conversion was obtained when increasing the H2/DCM molar ratio up to 100. Higher values did not lead to a significant increase in DCM conversion.

Effect of H2/DCM molar ratio on DCM conversion and selectivity to reaction products at different temperatures
A similar product distribution was also observed without significant changes in the selectivity to reaction products at all tested values of H2/DCM molar ratio.
4. Conclusion
Activated carbon-supported catalysts containing Pt nanoparticles were prepared and were demonstrated to be effective in the gas-phase hydrodechlorination of DCM at the operating conditions tested (∼250°C, 1 atm). The properties of the support play an important role in the dechlorination activity and stability of the catalysts. Whereas Pt/CE and Pt/CM maintained their activity during 26 days on stream, Pt/CC suffered rapid deactivation. In addition to their small Pt particle size and the high proportion of Pt0/Ptn+ ratio, the high stability of the former can be related to the lower superficial acidity of the activated carbon, since the acidity of the support favours coke deposition, causing deactivation of the catalysts. Increasing metal loads to 2% (w/w) led to an increase in Pt0/Ptn+ ratio while dispersion remained constant, thus maximizing the dechlorination activity of the catalyst. The effectiveness of the catalyst (tested Pt/CE) was maintained in a feed DCM concentration of 1000–3500 ppmv. The catalyst was active in the interval of H2/DCM molar ratios 50–200; however, a remarkable increase in dechlorination activity was obtained up to a value of 100, whereas no significant increase was observed for higher H2/DCM molar ratios.
Footnotes
5. Acknowledgements
The authors gratefully acknowledge financial support provided by the Spanish Ministerio de Economía y Competitividad (MINECO) through the project CTM2014–53008-R.