
Abstract
We have used density functional theory (DFT) and time dependent (TD)-DFT to systematically investigate the dependency of the geometric and vibro-electronic properties of zigzag and armchair-type doublewalled boron nitride nanotubes ((0,m)@(0,n) and (m,m) @(n,n)-DWBNNTs) on the interwall distance (ΔR) and the number of unit cells. The results of the calculations showed that their structural stability strongly depends on the interwall distance, but not on the number of unit cells, and the (0,m) @(0,m+9/10) and (m,m) @(n,n) with n=m+5/6 are the most energetically stable structures. The predicted electronic structures for DWBNNTs with cell lengths of one unit exhibit a strong red-shift for the ΔR below ~0.4 nm and remain almost constant for the ΔR > 0.45 nm. The calculated nonresonance Raman spectra of (0,6) @(0,n)-DWBNNTs (with cell lengths of one unit and n=12–18) indicated that the radial breathing modes (RBMs) of inner (0,6) and outer (0,n) tubes are not only diameter dependent, but also exhibit a strong blue-shift for the ΔR below ~0.35 nm and rapidly approach zero with increasing ΔR reference to the position of the RBM in the spectrum of the corresponding single wall boron nitride nanotubes, (0,n)-SWBNNTs. The calculated IR spectra of the (0,6) @(0,n)-DWBNNTs did not indicate any significant dependence on the ΔR for n > 13.
1. Introduction
Since the discovery of carbon nanotubes in 1991 by Iijima [1], they have received much interest in several fields due to their near-perfect geometric structure with almost endless applications in new technologies such as one dimensional quantum wires [2], transistors and sensors [3], in heat conduction systems [4], in specialty electronics [5], molecular memories [6], optics [7], photonic devices [8, 9], optical switches [10] and passively mode-locked lasers [11], electrically excited single-molecule light sources [12, 13], high-performance adsorbent electrode material for energy-storage devices [14], and protein functionalization [15]. Carbon nanotubes may have properties of either metals or semiconductors depending on their geometry [16], giving them many potential applications in new devices. Carbon nanotubes (CNTs) with small-diameters reveal many unusual properties such as anisotropic optical absorption spectra [17] and superconductivity originating from a Peierls distortion [18, 19]. These findings have motivated much interest in the study of small nanotubes both theoretically and experimentally [20–22].
Compounds with a propensity for graphite-like layering are natural candidates for nanotube formation, and indeed transition-metal dichalcogenides and hexagonal boron-nitride based nanotubes have been predicted [23] and observed in various studies [24–26]. Importantly, some of the predicted properties of noncarbon nanotubes are, for certain applications, superior to those of pure carbon nanotubes. For instance, boron nitride nanotubes (BNNTs) are resistant to oxidation up to 1100 C [27, 28], have excellent piezoelectricity [29], are good electrical insulators at room temperature [30] and have a potential hydrogen storage capability [31]. Moreover, because of their ionic bonding properties, BNNTs are wide band gap materials like other nitride materials and possess electronic properties that are not sensitive to a change in their diameter, chirality and number of tubular walls [19, 23, 32]. The cathodeluminescence (CL) of an individual MWBNNT exhibits strong band(s) at ∼5.5 eV [23]; ∼3.9 eV and a weak peak at ∼5.3 eV [33]. Later, optical absorption measurements showed that BNNTs might have an optical band gap at ∼5.9 eV and two sub-band absorptions at ∼4.8 eV and ∼3.7 eV [34]. The optical band gap of high-quality BNNTs was recently found to be ∼5.9 eV without sub-band absorption [30]. Owing to their wide band-gap, pristine BNNTs are insulators [30]. However, it was found that the carbon doping [35] and bending of an individual BNNT [36, 37] could convert the insulating behaviour into that of a semiconductor. The experimentally measured interwall distance (ΔR) between the inner and outer tubes of the DWBNNTs using high-resolution transmission electron microscopes (HRTEM) are reported as ∼0.35 nm by Ghassemi et al. [38], 0.36 ± 0.03 nm by J. Cumings and A. Zettl [39] and between 0.38 and 0.42 nm by R. Arenal et al. [40]. These measured values of the ΔR are larger than the 0.333 nm interlayer spacing of the flat hexagonal BN (h-BN) [41]. Of course, this is to be expected since the number of layers becomes larger, leading to an increase in the total interaction energy between them relative to a couple of sheets.
As is well known, the optical properties of nanotubes are implicitly connected with the absorption, photoluminescence and Raman spectroscopy of nanotubes. Such optical measurements permit a reliable characterization of the quality of nanotubes, such as by their chirality, size, and structural defects. The Raman spectrum of a nanotube exhibits a few characteristic modes that can be used to determine the size of nanotubes and to classify the type of the nanotubes. For example, in the low frequency region, one type of characteristic vibration is called the radial breathing mode (RBM), which is strongly diameter-dependent and is used to determine the size of the nanotube [16, 42, 43]. Additionally, there are two other characteristic Raman bands in the high frequency region of the Raman spectrum, which are called the tangential mode (TM or GM) and the disorder induced mode (DM). The line shape of the TM modes may be used to classify whether the nanotube is metallic or semiconducting. The DM is also important for defects on the nanotube surface as a consequence of chemical functionalization or of structural deformations. These Raman modes in the high energy region are also slightly diameter dependent. Several researchers have measured the Raman and Fourier transformed infrared (FTIR) spectra of the BNNTs [34, 44–46]. While the resonance or pre-resonance Raman spectrum of the BNNTs displays a strong peak in the range of 1361–375 cm−1, the FTIR spectroscopy revealed three IR peaks, namely, a relatively weak one in the range of 1510–1545 cm−1, a strong IR peak in the range of 1360–1375 cm−1, and two weak peaks in the range of 815 and 800 cm−1.
In our previous papers [16, 42, 43], density functional theory (DFT) with the split valence-type basis set was used to calculate the spectroscopic and geometric properties of SWCNTs/SWBNNTs and DWCNTs/DWBNNTs in order to examine the changes that occur when the system changes from SWNT to DWNT at the same level of the theory, B3LYP/6-31G. Since the DWBNNTs are on the border between the SWBNNTs and MWBNNTs, it is worth investigating the dependence of the structural and spectroscopic properties of the DWBNNTs on the interwall distance and the length of the nanotubes in more detail. In this study, as a continuation of our previous works, we investigated the dependence of the energetic stability and the spectroscopic properties of the DWBNNTs on the interwall distance and length of the tube for the (0, m)@(0, n)-DWBNNTs (n = 6-8 and n = 12-20) and (n, n)@(m, m)-DWBNNTs (n = 3-4 and m = 7-11). The geometric and spectroscopic properties of the boron nitride nanotubes were calculated at the B3LYP/6-31G level of the DFT as contained in the Gaussian 03 software package [47].
2. Methods
All nanotubes were optimized to minima on the potential energy surfaces without any symmetry restriction, which were verified through frequency calculations revealing no imaginary frequencies. In the optimized geometries of DWBNNTs, (0,m)@(0,n), (m,m)@(n,n), we placed each B (or N) atom in the inner nanotube so as to face a N (or B) atom in the outer nanotube, leading to the AB stacking order between the inner and outer tubes as in the case of the hexagonal stacking in the h-BN solid. The Raman intensity (I
Furthermore, for the (0,6)@(0,n)-DWBNNTs (n = 12 to 16), the time-dependent density functional theory at the TD-B3LYP/6-31G level was applied to investigate the dependence of the dipole allowed and forbidden vertical electronic transitions on the interwall distance. In the calculations, the DFT method was chosen because it is computationally less demanding than other approaches with regard to the inclusion of electron correlation. Moreover, in addition to its excellent accuracy and favourable computational expense ratio, the B3LYP calculation of Raman frequencies has shown its efficacy in numerous earlier studies performed in this laboratory and by other researchers, often proving itself the most reliable and preferable method for many molecular species of intermediate size, including anions and cations [52,53]. It may be worth noting that the calculated naturel orbital bond (NBO) analysis showed that the contributions from the N and B atoms to the formation of each Σ and π bonds are not equal:

In order to describe the change in interwall interaction energies between the inner and outer tubes of DWBNNTs as a function of interwall distance, we used various types of well-known equations (for the intermolecular interaction energies). The exp-6 potential function [48] is one of the best fits to the five sets of the calculated interwall interaction energies at the DFT level (see Figure 1), which is given by Eq. 1:

Calculated dependency of the formation energies per atom (in meV) on the interwall distance (ΔR) and the number of unit cells (or tube length) for the zigzag and armchair DWBNNTs. (A): (0,6)@(0,n) with n ranging from 12 to 20 and k=1, 2 and 3 unit cell lengths, (B): (0,m)@(0,n) with m ranging from 6 to 8 and n ranging from 12 to 20, and cell lengths of one unit, (C): (3,3)@(n,n) with n ranging 7 to 11 and k= 2 and cell lengths of four units, (4,4)@(n,n) with n ranging from 7 to 11 and cell lengths of two units, (D): change in the radius (r) and unit cell length (L) of the inner and outer tubes of (0,6)@(0,n) with n=12 to 20 and one unit cell length with reference to their corresponding (0,6)- and (0,n)-SWBNNTs, Δr and ΔL, respectively. It should be noted that the calculated formation energies for each of the DWBNNTs considered here are plotted in the figures A-C, but, in order to provide a better view of the energetic stability of the DWBNNTs, the data for the interwall distance (ΔR) below 0.30 nm are not displayed in the figures.

where Φ12(r) is the intermolecular_pair_potential between two particles or sites; r is the distance between two particles (=|r1-r2|); the σ is the value of r at which Φ12(r)=0; ε is the well depth (energy); and α is the steepness of the repulsive energy. The fitting equation reproduced the calculated formation energies at each interwall distance which were almost the same as those obtained at the DFT.
3. Results and discussions
Figure 1 provides the calculated formation energies (interwall interaction energies) in meV of the zigzag- and armchair-DWCNNTs. The formation energies (per atom) of the (0, n)@(0, m)-DWBNNTs (with n = 6 to 8 and m = 12 to 20) and (m, m)@(n, n)-DWBNNTs (with m = 3 to 4 and n = 7 to 11) were obtained from their calculated global energies relative to their corresponding isolated SWBNNT using the following equation:
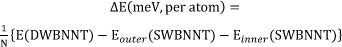
where E(DWBNNT), Eouter(SWBNNT) and Einner(SWBNNT) represent the calculated global energies of the DWBNNT and corresponding isolated SWBNNTs, respectively, and N indicates the total number of atoms in the DWBNNT (which is equal to the total number of atoms in the SWBNNTs). The results of the calculations indicate that the energetic stability of the DWBNNT strongly depend on the interwall spacing between the inner and outer tubes, but the dependence on the length of the nanotube is insignificant. Furthermore, as seen in Figure 1, since the predicted formation energies per-atom for the zigzag-DWBNNTs are about −4.1 meV more stable than the armchair-DWBNNTs, consequently, the results of the calculations suggested that the zigzag-DWBNNTs may be more easily formed than the armchair-DWBNNTs. Of course, this is to be expected since only two of three sp2 hybridized sigma bonds within the zigzag tubes are strained (or altered from planarity), in the armchair tubes, all the sigma bonds are strained.
The predicted formation energies of (0, m)@(0, n)-DWBNNTs (with cell lengths of one, two and three units) showed that (0, m)@(0, m+9)-DWBNNTs is only 0.47 meV above the most energetically stable structure of (0, m)@(0, m+10)-DWBNNT; thus, both structures are energetically favourable within a sample (Figure 1A-B). The average values of the predicted interwall distance (ΔR) between the inner and outer tubes are respectively 0.3603 and 0.3972 nm for (0, m)@(0, m+9)- and (0, m)@(0, m+10)-DWBNNTs. For (m, m)@(n, n)-DWBBNTs (with cell lengths of two and four units), the most energetically stable structure is (m, m)@(m+6, m+6)-DWBNNT (with ΔR = 0.3471), but there is a another favourable structure, (m, m)@(m+5, m+5)-DWBNNT (with ΔR = 0.4169 nm), is 1.5 meV above the most energetically stable structure (Figure 1C). The predicted values of the ΔR for the zigzag- and armchair-DWBNNTs are in good agreement with the experimentally measured values of 0.36 ± 0.03 nm determined by J. Cumings and A. Zettl [39], and 0.38–0.42 nm determined by R. Arenal et al. [40] for the DWBNNTs, and ∼0.35 nm for MWBNNTs by Ghassemi et al. [38].
The solid curves in Figure 1A-C are fitted to the calculated formation energies of the DWBNNTs using the exp-6 potential function [48] (in order to describe the interaction energy between the inner and outer tubes). The lowest point of the curves which gave average values of the ΔR are ∼0.377 nm for the zigzag-DWBNNTs and ∼0.379 nm for the armchair-DWBNNTs. However, the predicted values of the ΔR are much larger than the interlayer distance in h -BN (0.333 nm) [41], which may result from the fact that global interactions between the large number of layers in the h-BN is greater in the case of MWBNNT constructed by a few concentric tubes. Additionally, we also calculated the change in the radii and the lengths of the inner and outer tubes for (0,6)@(0,n)-DWBNNTs referenced to their corresponding isolated SWBNNTs, which are plotted as a function of the interwall distance (ΔR) as seen in Figure 1D. The results of the calculations showed that while the radius of the inner tube decreases, the length of the tube increases with decreasing interwall distance, and so both of them approach zero after ΔR > 0.35 nm. In contrast, with the outer tube, while the radius increases as ΔR increases, the length decreases, so that both of them rapidly approach zero after ΔR > 0.35 nm.
Calculated Electronic Spectra: Figs. 2(A-B) and 3(A-B) provide the calculated vertical dipole the allowed and forbidden electronic spectra of the zigzag- and armchair-DWBNNTs (with cell lengths of one unit) and their corresponding isolated SWBNNTs. The results of the calculations clearly indicated that all of the electronic transitions of the zigzag-DWBNNTs are significantly red-shifted relative to isolated zigzag-SWBNNTs. For instance, for the most energetically stable structures of the (0, 6)@(0, 16), (0, 7)@(0, 17), and (0, 8)@(0, 18) DWBNNTs, the calculations indicate that the lowest dipole allowed electronic transitions is due to the S0→S3 (4.62 eV; n → π*), S0→S7 (5.45 eV; n → n → π*) and S0→S2 (5.63 eV; n → π*), respectively. However, for the isolated (0, 6), (0, 7), (0, 8), (0, 16), (0, 17), and (0, 18) SWBNNTs, the lowest dipole allowed electronic transitions are predicted at: S0→S8 (5.89 eV), S0→S9 (5.84 eV), S0→S6 (6.03 eV), S0→S4 (5.91 eV), S0→S4 (5.91 eV), and S0→S4 (5.91 eV), respectively. The plot of the calculated electron densities of the DWBNNTs showed that these transitions result from the outer-shell to the inner-shell of the DWBNNTs. This finding clearly indicates the existence of the charge transfer from the outer-shell to the inner-shell via electronic excitation. Furthermore, as shown in Figure 2, many dipole allowed electronic transitions below 6 eV (as a result of the n → π* or n + π → π* transitions) are caused by the transitions from the outer (or inner) tube to the inner (or/and outer) tube(s) of the zigzag-DWBNNTs.

The calculated dipole allowed (red filled circle) and forbidden (black open circle) vertical electronic transitions: (A) as a function of interwall distance (ΔR) for (0,m)@(0,n)-DWBNNTs with m ranging from 6 to 8 and n=12 to 18 and one unit cell length; (B): as a function of tube radius (R) for (0,n) SWBNNTs with n ranging from six to 20 and cell lengths of one unit. The solid curves show that the calculated allowed electronic transitions exhibit interwall dependences that can be a fit for the exp-6 function (as given by Eq. 1).
Figure 2A provided that the dependence of the dipole allowed electronic transitions on the interwall distance (ΔR) between the inner- and outer-shells. As seen in Figure 2B, the first dipole allowed electronic transitions of (0,n)-SWBNNTs (n=6–20), which are predicted at around 5.9 eV, and almost no dependence on the tube radius, but the upper electronic transitions linearly change as a function of tube radius. However, the first dipole calculated allowed electronic transitions for the (0, 6)@(0, n)- DWBNNTs (n=12 to 20), relative to those in their corresponding isolated SWBNNTs (∼5.9 eV), which are lowered by as much as 1.3 eV for the ΔR < 0.4 nm, 0.6 eV (at ΔR =0.436 nm) and 0.1 eV (at ΔR =0.476 nm). For the ΔR > 0.5 nm, the predicted amount allowed electronic transitions of the zigzag-DWBNNTs and their corresponding SWBNNTs are almost the same, which is not so surprising since the formation energies (or the interaction energy between the inner- and outer-shells) of the DWBNNTs for the ΔR > 0.5 nm approach zero as discussed above. Furthermore, we found that the calculated amounts allowed electronic transitions at the TD-DFT level to exhibit interwall dependences that may be a fit for the exp-6 function (as given by Eq. 1).
Moreover, it was predicted that the lowest dipole the forbidden electronic transitions for (0, n)-SWBNNTs are 4.72, 5.05 and 5.61 eV for n = 6, 7, and 8, respectively, and about 5.8 eV for n = 9 to 20. For the (0, m)@(0, n)-DWBNNTs (m=6 to 8 and n=12 to 20), the lowest forbidden electronic transitions are predicted from 4.35 to 4.62 eV for the interwall distance ranging from 0.253 to 0.555 nm. Consequently, the forbidden transitions of the (0, m)@(0, n)-DWBNNTs are also ∼1.2 eV red-shifted relative to the lowest forbidden transitions for the (0, n>7)-SWBNNTs.
The calculated electronic spectra of the armchair-DWBNNTs exhibiting many dipole allowed electronic transitions below 6 eV. In the comparison between these transitions with the predicted allowed transitions of their corresponding SWBNNTs, as seen in Figure 3A-B, they also are red-shifted because of the intratube interactions. The calculated electron densities in some of the upper molecular orbitals (HOMOs) and lower unoccupied molecular orbitals (LUMOs) showed that these allowed transitions involve the transitions from one of the shells to not only one of the inner- and outer-shells, but also transitions to both shells. Therefore, for the armchair-DWBNNTs, there is evidence of a partial charge transfer mechanism. For instance, for the most energetically stable structures of the (3,3)@(9, 9)-DWBNNTs, the lowest dipole allowed electronic transitions (S0→S4) at 5.53 eV is caused by the transition from the only inner tube to inner or/and outer tube(s). The second lowest transition (S0→S6) at 5.79 eV is caused by the transitions from the outer tube to the outer or/and inner tubes. For the most energetically stable structures of the (4,4)@(10, 10)-DWBNNTs, the predicted lowest dipole allowed electronic transition (S0→S2) at 5.57 eV is due to the transition from the only inner tube to inner or/and outer tube(s). The second lowest transition (S0→S8) at 5.8 eV is due to the transitions from the only outer tube to the outer or/and inner tube(s). However, the first and second lowest allowing electronic transitions are predicted: at 5.79 eV (S0→S4) and 5.99 eV (S0→S5) for the (3,3)-SWBNNTs; at 5.67 eV (S0→S2) and 6.03 eV (S0→S9) for the (4,4)-SWBNNTs, and at 5.83 eV (S0→S2) and 5.93 eV (S0→S5) for the (9,9)-SWBNNTs. Therefore, the intratube interactions leading to red shifts in the dipole allowed electronic transitions for the (3,3)@(9, 9)- and (4,4)@(10, 10)-DWBNNTs are respectively ∼0.26 and ∼0.1 eV, referenced to the first and second lowest allowed transitions in their corresponding SWBNNTs. These red shifts in the allowed electronic transitions of the armchair-DWBNNTs are not as strong as those for the zigzag-type DWBNNTs. Furthermore, the dipole allowed electronic transitions of the armchair-DWBNNTs also significantly depends on the interwall distance (ΔR) below 0.4 nm, as seen in Figure 3A-B. When compared with the predicted spectra of the zigzag- and armchair-DWBNNTs, the red-shift in the electronic spectra for the zigzag-type DWBNNTs are not only greater than those in the armchair-DWBNNTs, but also the zigzag-DWBNNTs exhibited a strong charge transfer process.

Calculated dipole allowed (filled circle) and forbidden (open circle) vertical electronic transitions: (A) as a function of interwall distance (ΔR) for (m,m)@(n,n) (with m ranging 3 to 4 and n=7 to 11, and one unit cell length), (B): (n,n) SWBNNTs (n =3 to 10) as a function of the tube radius.
The results of the calculated electronic transitions are consistent with the experimentally measured absorption spectra of the boron nitride nanotubes. For instance, C. H. Lee et al. [34] have measured the absorption spectrum of the suspension of BNNTs in ethanol by using UV-visible absorption spectroscopy (HP 8453 Spectrophotometer). The authors observed three absorption bands at ∼5.9 eV (very strong) and ∼4.78 eV (weak), and ∼3.7 eV (very weak) in the UV-visible spectrum. They suggested that the band at about 4.75 eV originates from the intrinsic dark exciton absorption band; the relatively small band at ∼ 3.7 eV was due to the defects of the boron nitride nanotubes (BNNTs), and the stronger band at 5.9 eV was a result of the optical band gap of the BNNTs. These measured absorption bands at about 4.78 eV and 5.9 eV are very consistent with the calculated dipole which allowed transitions at 4.62 eV (S0→S3; n → π*) and 5.9 eV (S0→Sk>25; n → π*), respectively, for the (0,6)@(0,15/16)-DWBNNTs, although there is no evidence for the observed band at ∼3.7 eV. This transition might result from the defect on the surface of the nanotube as mentioned in reference [34].
Calculated Raman Spectra: it is well known that while the Raman spectra of the single-wall nanotubes exhibits a RBM (radial breathing motion) mode [16, 43], the double-wall nanotubes exhibited two RBM modes (resulting from the radial breathing motion of the inner- and outer-shells) in the low frequency region, and so both of the RBM modes are blue-shifted when compared with their spectral position in the spectra of corresponding isolated single wall nanotubes in the low frequency region [43]. In our previous works [42, 43], we calculated nonresonance Raman spectra for the (0, n)-SWBNNTs (with n ranging from 6 to 9) and (0, n)@(0, 2n)-DWBNNTs (n = 6 to 9). The calculations showed that the frequencies of the RBMs and tangential modes (TMs, known as G-mode) of (0, n)@(0, 2n)-DWBNNTs significantly differ from those calculated for the (0,n)-SWBNNTs. As a continuation of our previous works, in this work, we systematically studied the dependence of the Raman bands on the interwall distance for (0, 6)@(0, n)-DWBNNTs with n = 12–18. For the armchair-DWBNNTs, due to technical difficulties, we could only calculate the Raman spectra of (3, 3)@(m, m)-DWBNNTs, m = 7–9. Therefore, here we only discuss the results of the zigzag-DWBNNTs.
The calculated Raman spectra of (0, 6)@(0, n)-DWBNNTs indicate that there are two Raman bands, RBM (radial breathing motion) and ED (elliptical deformation) modes, in the low frequency region. The vibrational frequencies of these bands are not only found to depend on the DWBNNTs diameter, but also strongly depend on the interwall distance below 0.4 nm as shown in Figure 4A. Figure 4B provides the plot of the relative shift in the RBMs and ED modes of frequencies as a function of interwall distance (ΔR), referenced to their spectral position in the spectra of corresponding isolated SWBNNTs; Δωi = ωi(DWBNNT) - ωi(SWBNNT). As seen in Figure 4B, the RBMs (with A1 symmetry) and EDs (E2) modes of frequencies for the inner- and outer-shells rapidly decreases with increasing interwall distance and approaches zero at an interwall distance of nearly 0.4 nm corresponding to the energetically more stable structure, (0, 6)@(0,16)-DWBNNTs. For interwall distances smaller than 0.4 nm, the calculations also indicate that the dependence of the RBMs and EDs of inner-shells in the distance are stronger than the outer-shells of the zigzag-DWBNNTs. We obtained a best fit to the calculated data by using the first derivative of the Eq. 1 (exp-6 function).

(A): the calculated Raman spectra of (0,6)@(0,n) DWBNNTs (with n ranging from 12 to 18 and one unit cell length); (B): the plot of the change in the RBMs and DMs of the frequencies of the inner and outer tubes (reference to their corresponding frequencies in the (0,n) SWBNNTs) as a function of the interwall distance (ΔR); and (C): the plot of the Raman bands (TMs and DMs) of the frequencies in the region of high energy as a function of the ΔR. The A1, E1 and E2 indicate the symmetries of Raman modes, and the letters i and o in parenthesis indicate that the vibrations are due to the nuclear motions of the inner and outer tubes.
This fitting equation reproduced the calculated relative change in the RBMs within minute error.
In the region of the tangential modes (TMs) and disorder modes (DMs), the calculations exhibited a few Raman bands with the E1, E2 and A1 symmetries in the spectra of the DWBNNTs. For the most energetically stable structure of zigzag DWBNNT, (0,6)@(0,16)-DWBNNT, the vibrational frequencies and assignments of these bands are summarized as follows. The Raman bands with E symmetry: the Raman bands at 1265 (E1) and 1269 (E2) cm−1 are due to the asymmetric vibrational stretching of the NBN bonds of the outer-shell only. The band at 1288 (E1) cm−1 results from the NBN bonds stretching in the inner-shell only. The bending deformation of the NBN (or BNB) bonds and relatively weak BN bond stretching along the circumference direction are responsible for the predicted Raman bands at 1352 (E1) cm−1 (mostly caused by the outer-tube) and 1359 (E1) cm−1 (primarily caused by the inner tube). The Raman bands at 1411 (E1) and 1443 (E1) cm−1 result in asymmetric stretching of the NBN bonds and bending deformation of both the inner- and outer-shells. The bands at 1449 (E2) cm−1 are due to the asymmetric stretching and bending deformation of the NBN bonds of the outer-shell (mostly). The Raman bands with the A1 symmetry: the bending deformations of the NBN bonds along the tube axis and in the same phase are responsible for the Raman bands at 1331 cm−1 (inner tube only) and 1362 cm−1 (outer tube only). The Raman bands at 1464 and 1511 cm−1 are a result of the BN bond stretching (along tube axis and in the same phase) of both the inner- and outer-shells, which also cause the bending deformation of the NBN bonds. It is worth noting that even though the calculated nonresonant Raman spectra of the boron nitrite nanotubes exhibited a few Raman bands in the region of the TMs with the A1, E1 and E2 symmetries and two almost overlapping DMs with the E1 and E2 symmetries below 1300 cm−1, at resonance or pre-resonance condition, the TMs at around 1360 cm−1 are extremely enhanced and overwhelmed other Raman features in the spectrum as observed experimentally [34, 44–46]. Furthermore, Obraztsova et al. [50] have measured the Raman spectrum of the Al-modified MWBNNTs sample. Their measured Raman spectrum exhibited a broad feature around 1293 cm−1 in addition to the well-known Raman band at 1366 cm−1, known as TM, in the spectra MWBNNTs. The authors concluded that the TM at 1366 cm−1 and a broad band at ∼1293 cm−1 shows that the samples are formed by two BN phases: crystalline and amorphous-like. However, our calculations suggest that this broadband (at ∼1293 cm−1) might result from the defect modes (DMs) of the MWBNNTs, as predicted in the region of 1265–1288 cm−1 for the DWBNNTs.
The dependence of the TMs and DMs on the interwall distance is shown in Figure 4C. The calculations indicated that the TMs rapidly increase with increasing interwall distance (ΔR ≤ 0.35 nm) for the vibrational modes of frequency resulting in a pure or dominantly nuclear displacement of the outer tube, increasing with decreasing ΔR for the vibrations of pure displacement or displacement dominantly caused by the inner tube, and remain constant after the ΔR ≥ 0.35 nm. The DMs of the frequencies exhibited similar trends (see Figure 4C). These observations are consistent with the change in the radius, and the lengths of the inner and outer tubes as plotted in Figure 1D.
The calculated IR spectra: the calculated IR spectra of the (0.6)@(0.n)-DWBNNTs with n=12–18 exhibited few IR peaks, as seen in Figure 5A. The predicted IR peaks are at around 1442, 1412, 1348, 1258, 821 and 780 cm−1 with the E1 symmetry, and 1511 and 1362 cm−1 with the A1 symmetry. The experimental measured spectrum of the multi-walled BNNTs (MWBNNTs) revealed few IR peaks [34, 44–46]: at a broad and blue, it degraded strong IR peaks in the range of 1360–1375 cm−1 (with the full width at half of the maximum (FWHM) of about 100 cm−1), two relatively weak peaks at around 815 and 800 cm−1, and another relatively weak and broadened IR peak with the highest frequency was observed in the range of 1510–1545 cm−1. The IR peak measured at 1360–1375 cm−1 (FWHM= ∼100 cm−1) corresponds to the calculated IR peaks at 1442, 1411, 1365 and 1356 cm−1. The observed double peaks in the range of 815–780 cm−1 are consistent with the peaks calculated at 821 cm−1 (mostly caused by the buckling of the outer tube along the radial direction) and 781 cm−1 (mostly caused by the buckling of the inner tube). The calculated spectra of neither the DWBNNTs nor SWBNNTs produced an IR peak above 1515 cm−1. Consequently, the measured IR peak in the range of 1500–1515 cm−1 corresponds with the one predicted at 1511 cm−1, but the observed IR peak(s) above 1515 cm−1 might not belong to the BNNTs and may result from a combination of bands such as ca. 1511 cm−1 + (1443–1413) cm−1 =1541 cm−1. Furthermore, the dependency of the IR features on the interwall distance (ΔR) is shown in Figure 5B. The plot of the IR peaks as a function of ΔR indicated that for ΔR ≤ 0.32 nm. However, only the IR peaks in the range of 1400–1100 cm−1 depend on the ΔR. These findings indicate the existence of a strong repulsive interaction between the inner and outer tube as observed in the Raman spectra and formation energies of the DWBNNTs.

(A): the calculated IR spectra of (0,6)@(0,n) DWBNNTs with n ranging from 12 to 18 and cell lengths of one unit) and (B): the plot of the IR bands of the frequencies as a function of the ΔR. The A1 and E1 indicate the symmetries of the Raman modes.
4. Conclusion
We theoretically investigate the dependence of the geometric and spectroscopic properties for the zigzag and armchair DWBNNTs on the interwall distance (ΔR) and unit cell length using the DFT. Some interesting findings are summarized below:
The zigzag-type DWBNNTs are about 4.1 meV more stable than the armchair-DWBNNTs. The diameter (or radius) and the cell length of the unit of the inner and outer tubes DWBNNT alter with the interwall distance since the AR < ∼0.35 nm.
The predicted formation energies per atom of the DWBNNTs showed that their structural stabilities strongly depend on the interwall spacing, but dependence on the cell length of the unit is not significant. Even though it predicted that the most energetically stable structure of the zigzag-type tube is (0,m)@(0,m+10) with the ΔR=0.3972 nm, the second one (0,m)@(0,m+9) with ΔR=0.3603 nm is also energetically favourable since it lies about 0.47 meV above the (0,m)@(0,m+10). For the armchair tube, (m,m)@(m+6,m+6) with the ΔR=0.4169 nm is the most energetically stable structure, but the (m,m)@(m+5,m+5) with ΔR=0.3471 nm is predicted as 1.5 meV above the (m,m)@(m+6,m+6).
The calculated dipole allowed vertical electronic transitions of (0,6)@(0,15/16) are predicted at 4.65/4.62 eV (S0→S3; n → π*), which is ∼1.27 eV red shifted reference to the lowest allowed electronic transitions of their corresponding SWBNNTs. This red shift in the spectra of (0,7)@(0,16/17) are respectively ∼0.68 eV (S0→S4; n → π*) and 0.39 eV (S0→S7; n → π*); for (0,8)@(0,17/18), 0.34 eV (S0→S1; n → π*) and 0.28 eV(S0→S2; n → π*), respectively. Moreover, the calculated electron densities in the HOMOs and LUMOs showed the electron transfer from the outer tube to the inner tube.
The calculated nonresonance Raman spectra of (0,6)@(0,n) DWBNNTs showed that the RBMs of frequencies (reference to corresponding isolated SWBNNTs) are not only diameter dependent, but also depend on the interwall distance for the ΔR < ∼0.4 nm. The TMs and DMs of frequencies in the high frequency region have less dependence on the interwall distance since the ΔR > ∼0.35 nm. The experimentally well-known TM band, in the region of 1350–1370 cm−1, corresponds to three active Raman bands of symmetries A1(outer tube)+2E1(inner and outer tubes) that lie close to one another. The DM bands of symmetries E1(out), E2(out) at ∼1265 cm−1 and E1(inner) at 1288 cm−1 do not depend on the ΔR since ΔR >0.35 nm.
The experimentally reported IR peaks in the region of 815–780 cm−1 are consistent with the peaks calculated at 821 cm−1 (outer tube) and 781 cm−1 (inner tube), both have E1 symmetry which essentially remain constant. In the high frequency region, we predicted IR peaks at 1442, 1412, 1348, 1258 with the E1 symmetry, and 1511 and 1362 cm−1 with the A1 symmetry, which are in agreement with the experimentally reported IR spectra that exhibited a broad and blue degraded strong IR peak in the range of 1300–1540 cm−1.