
Abstract
Introduction
Almost 8,000 rare diseases exist worldwide, affecting approximately 350 million people. Nevertheless, only 5% receive a specific authorized or licensed treatment. The need for effective and rapidly available therapies is still unmet for many patients.
Objective
The objective is to define repurposing versus off-label drugs, and to evaluate pathways of repurposed drugs for rare non-oncological diseases in Italy, France, England, and Spain (the EU4 countries).
Methods
This original paper is based on 3 research activities: (i) a nonsystematic literature research; (ii) a questionnaire-based survey to regulatory experts; and (iii) research on approval timelines and therapy prices of repurposed non-oncology orphan drugs. Official approval dates in England are not available if the National Institute for Health and Care Excellence does not appraise the products.
Results
Only France provides a specific adaptive pathway from off-label to repurposed drugs. Pricing and reimbursement assessment for the drug samples varied across the EU4 countries: time-to-market for repurposed drugs versus new drugs is longer in all analyzed countries; that is, 979 days versus 462 days in Italy, 502 days versus 350 days in France, and 624 versus 378 days in Spain. Repurposed drugs have higher success rates from development to approval than novel drugs (30% vs. 11%).
Small- and medium-sized enterprises owned 9 of 12 repurposed non-oncology orphan drugs, of which only 4 were reimbursed in all EU4 countries. Prices were more homogeneous across EU4 although the reimbursement rates were different.
Conclusions
Drug repurposing represents a great opportunity to treat rare non-oncological diseases. However, a more homogenous assessment across EU4 could ensure reimbursement and prices high enough to reward organizations investing in this field.
Introduction
According to common definition, a disease is rare if it affects a limited number of people, which is no more than 5 out of every 10,000 people in the EU (1) and no more than 200,000 people or 1/1,250 in the USA (2).
Although each type of rare disease affects a limited number of people, the number of disorders that fit this definition is very large (up to 8,000) (3). Therefore, the number of patients amounts to around 350 million people worldwide, of whom 30 million are in Europe and 25 million in North America (4); this means that 1 in 17 people (7% of the population) will be affected by a rare disease at some point in life (5).
On top of this, only 5% of rare diseases have an approved treatment, even if specific treatments for rare diseases represent a consistent part of newly authorized drugs (41% of new drug approvals by the U.S. Food and Drug Administration and 16 out of 81 approvals by the European Medicines Agency's (EMA) in 2016 were for rare diseases) (6, 7). Indeed, only 112 orphan drugs were on the European market in July 2017 (8), of which almost half were for rare oncology disease, whereas more than 450 are under development (9). In this context, the need for effective treatments for rare diseases seems to become increasingly relevant.
The introduction of the Regulation on Orphan Medicinal Products in the European Union (EU) in 2000 (10) has been successful in making drugs for rare diseases commercially viable, giving incentives associated to orphan drug status, principally the 10-year market exclusivity. Authorized orphan medicines benefit from 10 years of protection from market competition with similar medicines that have similar indications, once they are approved. This period of protection is extended by 2 years for medicines that have also complied with an agreed pediatric investigation plan granted at the time of review of the orphan medicine designation. Beside market exclusivity, other incentives include protocol assistance, provision of scientific advice, access to the centralized authorization procedure, reduced fees for regulatory activities, some administrative and procedural assistance from the EMA's small- and medium-sized enterprises (SME) office, and opportunity to have access to grants from the European Commission and other sources (11).
As for any novel drug, once regulatory approval is obtained, manufacturers need to submit their application to the reimbursement authorities in each launch country. According to research on high cost orphan/nonorphan drugs (12), the time-lag between marketing authorization and reimbursement ranges from 7.0 to 11.2 months, with a minimal difference in time-to-reimbursement for oncology versus non-oncology drugs in the EU5.
For the next step, making an orphan drug available to patients requires further financial efforts and capabilities. While all drug launches are complex, launches of rare disease treatments are particularly so. A recent article (13) has highlighted that companies launching rare disease treatments not only must they show great commitment to the rare disease community, but also they need to identify patients and help patients and their caregivers navigate a health care system not usually geared to supporting those with rare diseases.
Overall, developing and delivering orphan drugs to patients requires high R&D and financial commitment, which makes this field difficult to be approached, especially by SMEs, in spite of EMA incentives. An opportunity for filling this gap is available by off-label use or repurposed (or repositioned) drugs. The latter option implies that costs of bringing repurposed drugs to the market are significantly lower than for novel medicines (14).
In this context, the aim of this work is to illustrate the reimbursement process and the regulatory status and prices of repurposed non-oncology orphan medicines in some European health care systems, starting from off-label regulations, in order to understand each different national approach.
Methods
This original paper was based on three activities: (i) questionnaire-based survey and expert opinion meeting; (ii) nonsystematic literature review; and (iii) research on approval timelines and prices of a selected sample of repurposed orphan drugs for non-oncological rare diseases in four European countries.
As a first step, finding an agreement on definitions related to drug repurposing and off-label use was necessary.
A questionnaire-based survey to 5 key regulatory experts from Italy, France, England, and Spain was administered and a relevant case study was analyzed, in order both to find agreement on definitions and to analyze the national contexts of drug repurposing. To maximize the effort, the interviewed experts were also invited for discussion purposes in a consensus meeting. Since no dedicated pathways exist in the countries of interest for repurposed drugs, the discussion was focused on off-label regulation country by country. The discussion focused on orphan medicinal products.
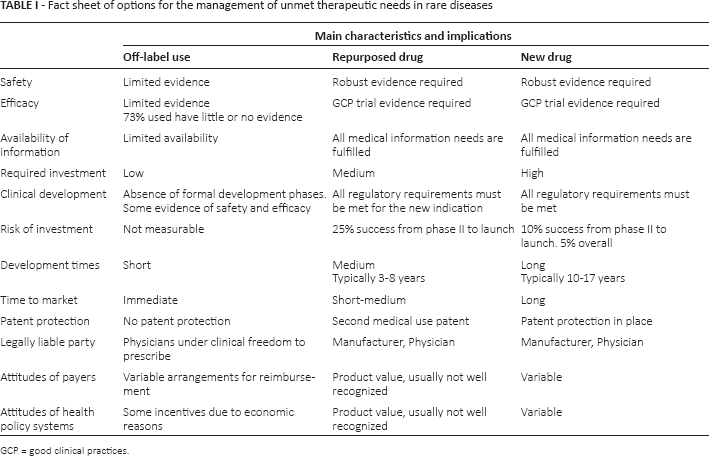
Second, a literature research on PubMed allowed us to fully understand the current standpoint on the subject, including the known definitions and the extent of the value that repositioning represents for such a sensitive subject as rare diseases. In order to determine the current regulatory standpoint on repurposed drugs (i) versus off-label use (ii) and new drugs (iii), these 3 statuses were compared for the following parameters: safety, efficacy, availability of information, required investment, clinical development, risk of investment, development times, time to market, patent protection, legally liable party, attitude of payers, and attitude of health policy systems.
The literature research included the key words “drug” AND “repurposed,” “repurposing,” “repositioning,” and “repositioned”; any article found relating to this subject was analyzed.
Finally, in order to put into effect the previously made considerations, non-oncology repurposed drugs – with an orphan designation and approved by the EMA – were collected, and the approval process and prices were analyzed country by country. Starting from the EMA's orphan drug list (8), we analyzed each drug with the aim of understanding whether they could fit the definition of “repurposed.” There were 112 currently approved drugs with an orphan drug designation; of them, 22 cases fit the definition of “repurposed” drugs. Of these, 12 were repurposed for non-oncological rare diseases, thus excluding oncological drugs.
A thorough search of each of these drugs was performed on the European and national authorities' websites: EMA, AIFA (Italian Medicines Agency), HAS (French High Health Authority), NICE (National Institute for Health and Care Excellence)/NHS (National Health System), and AEMPS (Spanish Agency of Medicinal Products and Medical Devices). The regulatory steps of each drug were tracked, and the date of the marketing authorization (EC decision) was used as the index date for comparison purposes. It was not possible to calculate timelines in England, because official approval dates are not available if NICE does not appraise the product. Finally, we described the price for reimbursed packages and the reimbursement status across the analyzed countries. Prices are shown as follows: ex-factory prices in Italy, the manufacturer price before taxes (PFHT) in France, the list price in England (converted to Euro according to the exchange rate as of July 21, 2017), and the public price minus 7.5% discount in Spain.
Results
Definitions
A recent review showed that no common definition for drug repurposing was identified in the literature. We referred to a definition given below (15-17), which was submitted and approved by all experts. The outcomes of the questionnaire-based survey and the expert opinion meeting are listed in this section as follows.
Orphan drugs
Drugs granted an orphan drug designation by regulatory agencies, intended for diagnosis, prevention, or treatment of life-threatening or very serious diseases or disorders that are rare (18). Currently, more than 1,800 orphan drug designations have been granted by the European Commission based on a positive opinion from the Committee for Orphan Medicinal Products (COMP) (7). Despite the evident promises of the orphan drug designation and the high medical need, there is still a considerable lack of special treatments on the market, due to the low return on investment and high developmental and financial risks (19).
Drug repurposing
Drug repurposing is the practice of finding novel therapeutic indications for existing drugs (15). It involves the R&D process and the application for regulatory approval of known drugs and compounds to treat new indications (i.e., new diseases) (16). Repositioning can give new life to shelved or abandoned drugs that have never been on the market, or it may extend the life for marketed drugs via new indications or formulations (17). Drug repurposing is not a simple re-use of a drug. Whilst the active ingredient is the same, dosages and/or the route of administration may vary considerably; hence the need to assess safety and efficacy of the repurposed drug.
Patent protection
Based on Article 54(5) of the European Patent Convention, a second or further medical use of a substance or composition can be so broad as to relate to “any specific use of such substance or composition in a therapeutic method.” Novel or further medical uses must not be chosen arbitrarily, but they must contribute to the claimed technical effect to be considered inventive (20). If a substance or composition is already known to have been used in a first medical use, it may still be patentable for any second or further use – subject to a number of exceptions – provided that said use is novel and inventive (20). Authorized orphan medicines benefit from 10 years of protection from market competition with similar medicines that have similar indications once they are approved (vs. 8 years data exclusivity +2 years market protection for nonorphan drugs). This period of protection is extended by 2 years for medicines that have also complied with an agreed pediatric investigation plan granted at the time of review of the orphan medicine designation (21).
Off-label drug use
An off-label drug is a drug used in a different way than that specified in the label (i.e., SmPC – Summary of Product Characteristics) covered by the authorization.
Concept overview
The results of the literature research on PubMed and a consultation with experts are summarized in Table I.
Fact sheet of options for the management of unmet therapeutic needs in rare diseases
GCP = good clinical practices.
In terms of safety and efficacy requirements, repurposed drugs are equivalent to newly approved drugs. Indeed, the safety and efficacy of repurposed drugs, as well as new drugs, need to be supported by clinical trials. On the contrary, the safety and efficacy of off-label use is limited to a few cases in clinical practice (generally case reports, case series, or small studies) (22).
All information on repurposed and new drugs are available and accessible through the European Public Assessment Reports (EPARs) and in the Summary of Product Characteristics (SmPC), published in the EMA website once authorized, whereas information on off-label use is limited to published and grey literature.
Off-label, repurposed, and new drugs require different levels of investment and development time. Development times are the longest for new drugs, typically 10-17 years, from in vitro studies to clinical studies with a very low percentage of success and, by consequence, high R&D costs: 10% success rate from phase II to launch and 5% overall (23). In the case of repurposed drugs, preclinical research and phase I trials are already available from the previously approved indication. This translates into reduced development times (3-8 years on average) and a higher probability of success than new drugs from phase II to launch (25% vs. 10%), leading to a reduction in the necessary investments. Comparing the number of approvals, repurposed drugs appear to be more frequently approved than novel drugs (30% vs. 11%) (23).
New drugs and repurposed drugs undergo the same “full approval” process, whereas off-label use - by definition -does not require formal approval from a national authority. The physician is the only liable party and can prescribe under clinical freedom (24). The legally liable parties in new and repurposed drugs are both the manufacturer and the physician.
No patent protection is provided for drugs used off-label, while new and repurposed drugs are covered by existing patent protection; the patent covers the new indication for repurposed drugs. Moreover, orphan drugs (thus including repurposed ones) are entitled to market exclusivity for a period of 10 years (or 12 years in cases of rare pediatric disease) (21).
A case-by-case assessment of the value of repurposed and new drugs is always required, and the attitude of payers and health policy systems in recognizing their value is variable.
Regulation of off-label use in EU4
Italy
In Italy, the law establishes that physicians can prescribe drugs only according to the therapeutic indications, regimens, and administration routes authorized by AIFA (art. 3, para. 1, Law 94/1998); thus, off-label use is not defined by law, but it is considered as every usage not indicated in the SmPC. The off-label use lies on 2 pathways: off-label use “for named patient” and “for drug.”
In the first case (for named patient), physicians may waive the ordinary rule that relates to the on-label prescription for the treatment of a single patient with drugs authorized for a different indication, or with a different route of administration, or different posology, or with a “magistral formula,” under their choice and liability, and in compliance with the conditions of the law. The physician believes that the patient cannot be adequately treated with the existing authorized alternatives; the off-label use is supported by accredited published scientific evidence - at least phase II trials; and the patient has been duly informed and has expressed their consent.
In the second case (for drug), for diseases that lack a dedicated therapy (unmet need), off-label prescription is possible also according to Law 648/1996 (Tab. II). Requirements are established in art. 1.4 regulating the off-label use “in the absence of a valid alternative” and cover innovative drugs authorized in other countries, unauthorized drugs still under clinical development (but at least with phase II results), and authorized drugs for unauthorized indications. Such a use is subject to the prior assessment of AIFA; once the AIFA Technical Scientific Committee (CTS) has approved the off-label use, the drug is included in a specific list and is reimbursed by the National Healthcare System. If the drug is not on the Italian market, the company is free to decide its price. The application for inclusion in the 648 list can be filed by CTS, patient associations, scientific societies, or universities.
Main characteristics of off-label prescriptions in Italy, Spain, France, and England

ATU = Autorisation Temporaire d'Utilisation; RTU = Recommandations Temporaire d'Utilisation.
Law n. 79/2014 has amended Law 648 introducing a new scenario under art. 1.4 bis: the off-label use “even in the presence of a therapeutic alternative” if the proposed drug is cheaper than approved products. In this event, monitoring tools (both for therapeutic and economic compliance) shall apply according to AIFA decision.
A specific public fund according to Law 326/2003 is available for the use in Italy of orphan drugs in rare diseases and for compassionate use in serious diseases during the time necessary for placing it on the market.
France
In France, off-label use is defined by the regulator, and the relevant importance is given to advocacy groups.
France is the only country among the EU4 analyzed to provide a specific adaptive pathway from off-label to repurposed drugs.
The ATU (Autorisation Temporaire d'Utilisation) is similar to the Italian Law 648, but is restricted to drugs with no previous market authorization. The price is free, but is not reimbursed. The difference between the price in ATU and the negotiated price will be reimbursed after drug authorization. Once market authorization is granted, ATU is switched to Law 48 until the completion of price negotiations (dispositif pérenne).
The RTU (Recommandations Temporaire d'Utilisation) is for drugs that have a label (different from ATU), but have to be used off-label. In this case, the medicine agency may grant a 3-year permission with mandatory implementation of a registry paid by the manufacturer. At the end of RTU, off-label use can be switched to full approval (Tab. II).
England
In England, an off-label medicine is defined as a medicine with an existing marketing authorization that is used outside its terms, and it is not expected that the existing marketing authorization will be extended to cover this use in the following 2 years. The off-label use can be requested by the clinician under his own responsibility. If an individual funding request is required, the clinical exception has to be proven for reimbursement. Usually, involvement by NICE is not required. For orphan drugs, there is usually only one single national payer. If the off-label use of a drug can be managed within the budget, no particular issues subsist, otherwise every case is judged separately (Tab. II). The physician usually prescribes the active ingredient and not the brand name, so hospitals can choose the most inexpensive alternative.
NICE provides Evidence Summaries on Unlicensed and Off-label Medicines (ESUOM reports), which are evidence reviews of the clinical data supporting the off-label use of a medicine. It is important to note that an ESUOM does not constitute an official guideline by NICE: the published reports remain advisory in contrast to the binding funding requirements attached to the Technology Appraisals and Highly Specialized Technologies programs, which appraise licensed treatments.
Spain
The structure of the Spanish health care system and its constraints are similar to those of the Italians. Nevertheless, in Spain, no laws concerning off-label use exist, but the general practice is that once an off-label use is granted for 1 patient, it is automatically approved for all subsequent patients.
There are 2 ways to obtain off-label reimbursement: by provider, usually represented by the hospital, or by regional administration. In the first case, the off-label use has to be a consequence of compassionate use. In the second case, off-label prescription should be notified to the Ministry of Health (Tab. II).
The considerations on off-label usage in Italy, France, England, and Spain are summed up in Table II.
Pricing and reimbursement process in EU4
Italy
After EMA marketing authorization, the pharmaceutical company may submit a pricing and reimbursement application to AIFA. Usually, C(nn) class is assigned within 1 month. The CTS evaluates efficacy, added value, innovation, and pharmacoeconomics; if added therapeutic value is given, the Pricing and Reimbursement Committee (CPR) negotiates price and reimbursability class with the company (25).
France
After EMA marketing authorization, the Transparency Committee (TC) performs the Health Technology Assessment (HTA). Together with the HAS, it provides an opinion to the Healthcare Product Economic Committee, which, in turn, decides on pricing issues; whereas the National Health Insurance decides on the level of co-payment (26).
England
EMA marketing authorization is reviewed by the Medicines and Healthcare Products Regulatory Agency (MHRA) and the pricing is agreed through the pharmaceutical price regulation scheme (PPRS). The PPRS is a voluntary, noncontractual agreement negotiated between the Government and the Association of the British Pharmaceutical Industry. It lasts 5 years and controls the pricing of all licensed, branded drugs sold to the National Health Service (NHS) throughout the UK. The aim of the scheme is to ensure that the NHS obtains drugs at a fair price while promoting a strong industry. Once pricing is agreed, NICE only appraises drugs referred by UK Ministers; however, if there is no referral, funding is based on local processes (27).
Spain
After EMA or AEMPS marketing authorization, the Directorate of Pharmaceutical and Health Products (DGFPS) evaluates efficacy, safety, and innovation, and decides whether the drug should be reimbursed. DGFPS and the Interministerial Pricing Committee (IPC) define pricing. Only when the drug is considered innovative, the company is involved in pricing negotiations (28).
Repurposed drugs approval timelines, prices, and reimbursement
From 112 orphan drugs with marketing authorization (8), 22 (20%) were repurposed or repositioned and 12 (55%) were non-oncological drugs for 10 different rare diseases (Tab. III). Three of these 12, based on cholic acid, had indications in inborn errors of primary bile acid synthesis.
List of sample drugs

EMA approval ranged from June 2007 for hydroxy-carbamide (brand name Siklos) to April 2017 for chenodeoxycholic acid (Chenodeoxycholic Acid Leadiant), implying that, for this recent approved drug, reimbursement dates were not yet available for any of EU4 health care systems. Except for Novartis, Shire, and Actelion, companies marketing these repurposed orphan drugs were SMEs (Tab. III).
Only 4 out of 12 drugs were marketed in all EU4 countries (macitentan [Opsumit]; cholic acid [Orphacol]; thiotepa [Tepadina]; everolimus [Votubia]) and 50% of them by SMEs. Three drugs (ketoconazole [Ketoconazole HRA]; hydrocortisone [Plenadren]; hydroxycarbamide [Siklos]) were marketed in 2 countries, and 66% of them by SMEs. Finally, 3 of 12 drugs were not reimbursed or reimbursement decisions were not yet available. Two of these included the recently EMA-approved chenodeoxycholic acid (Chenodeoxycholic Acid Leadiant) and cholic acid (Kolbam) (100% SMEs) (Tab. IVA, B). Cholic acid (brand name Kolbam by Retrophin Europe Ltd, previously owned by ASK Pharmaceuticals GmbH) was approved by EMA in November 2015 despite the General Court of the Court of Justice of the European Union on June 11, 2015 issuing its judgment in the case between Laboratories C.T.R.S. and the European Commission concerning market exclusivity for orphan medicinal products (Case T-452/14 (29)), and granting cholic acid (Orphacol) orphan market exclusivity. However, in our research, no local marketing authorizations have been found for this drug.
Approval times in four EU countries for repurposed orphan drugs for genetic non-oncological rare diseases

AIFA = Italian Medicines Agency; CT = Commission de la Transparence; EMA = European Medicines Agency; NICE = National Institute for Health and Care Excelence; P&R = Pricing and reimbursement.
Source:
Journal Officiel de la Republique Française.
Reimbursement of the analyzed repurposed orphan drugs

ASMR = Assessment of Improvement of Medical Benefit; CT = Commission de la Transparence; NHS = National Health Service.
Source:
Journal Officiel de la Republique Française.
Of 12 drugs, 9 were reimbursed in at least 1 of the EU4, but under different reimbursement schemes. In Italy, 6 of these drugs were reimbursed: ketoconazole, macitentan, cholic acid, hydrocortisone, thiotepa, and everolimus all received full reimbursement (class A or class H reimbursement designation), but none of the analyzed drugs was granted innovative status. In France, 5 of these 12 drugs were reimbursed or partially reimbursed. According to the French authority assessment, Assessment of Improvement of Medical Benefit (ASMR) grades differed greatly: only cholic acid (Orphacol) received a grade I ASMR designation (major improvement) with 65% reimbursement (not included in the ALD30, the list of long-lasting diseases for whom 100% reimbursement is granted). Everolimus was granted grade II (important clinical added value) and 100% reimbursement. Ketochonazole, hydrocortisone, hydroxycarbamide, and thiotepa received grade IV (minor improvement) and 65% reimbursement, except for thiotepa, where reimbursement status is not published. Macitentan was classified ASMR V and was not granted reimbursement (Tab. IVB).
In England, the only drug included in the list of nonreimbursed drugs through national prices is everolimus; thus, all other marketed drugs are reimbursed (Tab. IVB).
In Spain, 6 of these 12 drugs were reimbursed: macitentan, cholic acid, hydrocortisone, and thiotepa – being hospital drugs - were reimbursed at a 100% rate; whereas hydroxycarbamide (by Laboratorios Rovi, licensed by Marketing Authorization owner Addmedica) and everolimus – being prescription drugs – were reimbursed at a 60% rate.
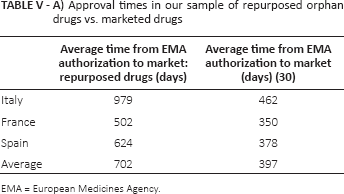
In Italy, France, and Spain, the average time-to-market from EMA approval to local market entry for novel drugs ranged from 350 days (11.7 months) in France to 462 days (15.4 months) in Italy (30). In these repurposed non-oncology orphan drugs, time-to-market varies from a minimum of 502 days (16.7 months) in France to a maximum of 979 days (32.6 months) in Italy (Tab. VA). However, we considered the following exceptional cases to be excluded from these calculations: cholic acid (Orphacol) in Italy and Spain (reimbursed after June 2015 Court of Justice sentence), and hydroxycarbamide (marketed in Spain by Laboratorios Rovi, outlicensed by Addmedica). Excluding these cases, approval times in repurposed drugs varied from 788 days (26.3 months) in Italy to 502 days (16.7 months) in France, and 365 days (12.2 months) in Spain (Tab. VB).
Approval times in our sample of repurposed orphan drugs vs. marketed drugs
EMA = European Medicines Agency.
Approval times in our sample of repurposed orphan drugs vs. marketed drugs (Orphacol and Siklos excluded)

EMA = European Medicines Agency
Analyzing prices for reimbursed packages, ex-factory prices are quite homogenous across the EU4 countries with some relevant differences in Spain for hydroxycarbamide (+44% vs. average EU4 price) and thiotepa (+15% vs. average EU4 price), and in Italy for cholic acid (Orphacol) (+13% vs. average EU4 price), with French prices falling in the lowest range (Tab. VI).
Package price of the analyzed repurposed orphan drugs

Ex-factory price.
PFHT.
List price, £ converted to €, according to exchange rate July 21, 2017 (1.117).
Public price minus 7.5% discount.
Discussion
There is an urgency to treat rare diseases. Although each of them affects only a limited number of individuals, taken together they strike approximately 30 million patients in Europe. Today, only 5% of rare diseases can be treated with an approved therapy; therefore, providing a rapid and effective solution for patients is an urgent need.
Novel drug development has relevant implications in terms of timing, development costs, and limited return on investments, making rare diseases not very attractive even for big pharma companies.
In this paper, with regard to the EU4 (Italy, France, England, and Spain), we examined the possible alternatives – off-label and repurposed drugs – which represent more rapid and more affordable solutions for pharma companies (especially small to medium ones) and for nonprofit organizations intending to develop drugs for rare diseases.
First, an agreement on definitions was required; and second - through a series of case studies – an analyses of regulations and reimbursement in these four countries, validated by experts' opinion, was performed.
Off-label use
Off-label use (a drug used in a different way than specified in the label covered by the authorization) has the advantage, in theory, of rapid availability of drugs that are already authorized in the country or in other countries. However, the regulatory framework in the EU4 on the subject of off-label drugs is varied. Italy and France have specific laws concerning the off-label use of drugs; namely, Law 648/1996 in Italy and ATU in France. France is also the only country to exhibit an adaptive pathway (RTU), at the end of which the drug may be switched to full approval. Off-label drug use is reimbursed according to Law 648/1996 in Italy; it is judged case by case in England, and is reimbursed by provider or regional administration in Spain. In England, NICE undertakes a small number of evidence reviews of the clinical data supporting the off-label use of a medicine. The published report remains advisory in contrast to the binding funding requirements attached to the Technology Appraisals program that appraises licensed treatments.
In summary, a nonhomogenous picture arises in the EU4 concerning regulations and reimbursement of off-label drug use, which does not allow this use to be approached in a systematic way to provide solutions to patients affected by orphan diseases, even though the International Rare Disease Research Consortium has set an ambitious objective to deliver 200 new therapies for rare diseases by 2020 (10).
Advantages and hurdles of repurposing
Repurposed drugs (off-patent, shelved, or abandoned drugs) appear to be a more viable option, as they overcome numerous challenges associated with the R&D of new drugs. These drugs have already been well studied and characterized in terms of preclinical toxicity, pharmacokinetics, safety, and processes for production. Few further data are required for a repurposed drug, mostly on safety and some pivotal trials. Bibliographic evidence is permitted to support market access (MA) application (31). Consequently, repurposed drugs come to market more quickly than new drugs (32-35).
Thalidomide is the most famous and well-documented case of drug repurposing, from the unfolding of devastating teratogenic effects to its current use as an effective treatment in leprosy, multiple myeloma, and other diseases (36). Thalidomide has captured the public imagination as a symbol of the dangers of inadequate research and regulation in pharmaceuticals and, after much careful research and investment, it has proven to be an example of the benefits of drug repurposing.
We focused our analysis on repurposed orphan drugs for non-oncology rare diseases, as a less homogenous group of drugs that also have a lower perception of life-threatening conditions.
Of these 12 identified cases, we were able to examine the path from EMA approval to reimbursement and agreed prices in their respective countries. While regulation is centrally ruled by EMA, with homogeneous requirements and approval timings in all EU countries, pricing and accessibility of orphan drugs work at the national level; they are often driven by HTA outcomes and with variable impact from external reference pricing. National pricing regulations are often value-based and the value placed on orphan drugs varies according to the health care system. In Sweden, Norway, and England, orphan drugs are freely priced at the manufacturer level, but are still subject to indirect regulations and profit control (22).
In a paper presented to ISPOR 2014 (12), average time-to-reimbursement in the EU5 after the EMA approval ranged from 7.0 to 11.2 months for high-cost orphan/nonorphan drugs. Also in our analysis, time-to-local-market is not homogenous across EU4, resulting in an even longer time overall. France showed a much faster process for repurposed non-oncology orphan drugs than the other EU4 countries, and was in line with time for new drugs, whereas in Italy and Spain, time from EMA approval to market is longer for these repurposed drugs than for new drugs. The delay in time-to-market in Italy and Spain is inconsistent with the advantages that drug repurposing brings about, and it could represent an obstacle for companies that investigate on this subject. Nevertheless, all of the approved drugs in Italy obtained full reimbursement by the National Health Service, while in France the reimbursement rate varied considerably.
On average, among the analyzed repurposed orphan drugs, hydroxycarbamide and thiotepa are the cheapest therapies, while cholic acid (Orphacol) and macitentan are the most expensive. Therapy prices depend on the number of patients as well as the availability of generic drugs. Indeed, cholic acid (Orphacol) and macitentan may be the most expensive therapy among those analyzed, targeting the diseases with the lowest prevalence. In contrast, the relatively low price for hydroxycarbamide and thiotepa might be justified by the available low-cost generics.
Hydroxycarbamide is a relevant case from this perspective. Hydroxycarbamide was an antineoplastic molecule that was commercialized as Hydrea in France from 1968. In the absence of an alternative licensed drug for the treatment of sickle cell syndrome (SCS), Hydrea has been routinely used off-label for over 15 years in this indication. In 2007, Siklos became the first licensed orphan drug indicated in the symptomatic treatment of SCS (37). However, despite the French TC accepting the high value of the product (high SMR granted) and recommending a 65% reimbursement, it decided to compare Siklos to Hydrea and, as a consequence, Siklos was evaluated as bringing a weak additional benefit (ASMR IV) (38), which had a major impact on the price given by the French Economic Committee on Health Care Products (CEPS). The manufacturer of Siklos entered in litigation with the CEPS, which eventually led the French Council of State to rule in this case and grant a higher price (although it was lower than English and Spanish prices) (39).
On the other hand, if marketed generics could be an opportunity (once a generic drug repurposing opportunity has been identified), they represent a relevant challenge on price, and thus a return on investments for those organizations intending to develop and market repurposed orphan drugs. Although the costs of bringing repurposed drugs to market are lower than new drugs (14), particularly after discounting cost of discovery (40), there are still costs that involve patent application, manufacturing, supply and distribution, promotion, and sales. Incentives like 10- or 12-year patent coverage are no doubt encouraging, but are not enough for small companies or not-for-profit organizations.
Companies involved
It is worth noting that many of the start-up companies investing in rare disease and/or repurposed drugs come from university spin-offs or even some patients' parents (i.e., Amicus Therapeutics, Lysogene). Furthermore, the rationale to develop repurposed drugs often comes from single case reports of off-label drug usage, with positive clinical results; therefore, they require more favorable legislation in this field.
Interestingly, most of these repurposed orphan drugs' MA owners were SMEs, but only 2 of them were able to market drugs in all EU4 countries (Laboratoires CTRS and Adienne). However, in some cases, licensing out has been a solution to overcome the relevant effort required to launch a new drug -even an orphan repurposed one. In fact, 2 out of 4 drugs marketed in all EU4 countries are launched by big pharma companies.
To promote repurposed orphan drug development and commercialization, a recent paper (10) has indicated interesting and factual business models for not-for-profit organizations. In our opinion, there is also a strong need to improve awareness on the subject, as product value is often not well recognized by payers and health policy systems. In fact, despite the clear benefits of repurposing for rare diseases, payers may regard companies focused on repurposing as similar to those producing generics, and will resist pricing that a sponsor may assume reflects the value of the product.
A defined reimbursement framework would be of great advantage both for companies who invest in repurposing – especially SMEs – and for patients to get easier and faster access to effective therapies. Specific regulations and detailed guidelines should be defined at both European and national levels.
For instance, when a positive safety profile is demonstrated, a repurposed orphan drug could be granted a centralized simplified procedure, such as the one in place for generic drugs. In addition, at the national level, a fast pricing and reimbursement procedure should be adopted, by defining a deadline from the approval date within which the price will be published.
Limitations of the study
The limit of our research is represented by the analysis of non-oncology orphan drugs, which does not allow us to draw definite conclusions on all repurposed orphan drugs. However, it has helped us to describe some situations that are often encountered with repurposed orphan drugs and to highlight the key issues in this field.
Conclusions
Providing a rapid and effective solution for patients suffering from rare diseases is an urgent need as only 5% of these diseases can be treated today by an approved therapy. Novel drug development has relevant implications in terms of timing, development costs, and limited return on investments, while for off-label use of authorized drugs, a nonhomogenous picture (in terms of regulations and reimbursement) arises in EU4, which does not allow this use to be approached in a systematic way to provide solutions to patients affected by orphan diseases.
Repurposing drugs represents a good opportunity to find effective treatments in rare diseases, offering advantages, such as shorter approval timings and lower investments. This emerging practice, which is often carried out by small companies, start-up companies, and not-for-profit organizations (e.g., research centers and universities) should be encouraged.
However, the regulatory status of repurposed non-oncology orphan drugs across Europe is deeply differentiated; for many of these drugs the national regulatory authorities have taken different decisions on pricing and reimbursement. A more homogenous assessment across the EU4 countries could ensure reimbursement and prices high enough to reward organizations investing in this field.
Footnotes
Acknowledgments
The article was written in collaboration with Arianna lorio, Dario Lidonnici, Stefania Niedecker, Virginia Ronco, and MA Provider.
Financial support: This publication was possible thanks to an unconditional grant from Santhera.
Conflict of interest: The authors declare no conflict of interest.
{kind=link}