
Abstract
Aims
Metabolic alterations in cancer, including bladder cancer, have been addressed in recent years. We aimed to study the role of phosphofructokinase (PFK) in muscle-invasive bladder cancer (MIBC).
Method
By in silico analysis of the bladder cancer data from the Cancer Genome Atlas (TCGA) database using the cBioPortal platform, we studied genetic alteration of genes within the PFK family (PFKL, PFKM, PFKP, PFKFB1, PFKFB2, PFKFB3, and PFKFB4). In vitro studies were carried out using the PFK inhibitor 2,5-anhydro-D-glucitol-6-phosphate.
Results
Genetic alterations of PFK family genes were observed in ~44% of MIBC cases in TCGA. The main alterations were amplification and upregulation. Patients with altered PFK gene status were more likely to have a history of noninvasive bladder cancer. Altered PFK status was not associated with survival or disease relapse. Use of the PFK inhibitor significantly decreased the level of glycolysis and inhibited the growth and invasion of bladder cancer cells.
Conclusions
PFKs were critical genes in charge of glycolysis and were upregulated in bladder cancer. Targeting this pathway could inhibit cell growth in bladder cancer.
Introduction
Bladder cancer, also known as urothelial carcinoma of the bladder, is a common malignancy in the genitourinary system and is characterized by multicentricity, high recurrence and potent invasiveness (1, 2). The necessity of lifelong surveillance has made bladder cancer one of the most costly diseases from diagnosis to death (3, 4). Despite the variety of current therapies for bladder tumors, including radical or palliative resection, chemotherapy, and radiotherapy, there is still an incorrigible rate of recurrence (5, 6). The recurrence rate of bladder carcinomas is up to 80%, of which 16%-25% face progression in grade and 10% become invasive (7-8-9).
Cancer adopts various types of metabolic rewiring, and the glucose metabolism is one of the most common energy shifts for cells to survive in the presence of hypoxia and overgrowth (10). Examples of this reprogramming in cancer include the well-established Warburg effect, in which glycolysis occurs constantly even in the presence of an affluent energy supply (10). Phosphofructokinase (PFK) is a kinase enzyme with a fundamental role in glycolysis. The enzyme-catalyzed transfer of a phosphoryl group from adenosine triphosphate (ATP) is an important reaction that occurs in many biological processes (10). PFK is one of the main enzymes that utilize this reaction: it catalyzes the phosphorylation of fructose-6-phosphate to fructose-1,6-bisphosphate, a key regulatory step in the glycolytic pathway.
The clinical significance of PFK can be classified into 3 groups: a) a genetic mutation in the PFKM gene results in Tarui's disease, a glycogen storage disease where the ability of certain cell types to utilize carbohydrates as a source of energy is impaired (11); b) some viruses affect cellular metabolic pathways such as glycolysis by a titer-dependent increase in the activity of PFK (12); for instance, herpes virus increases the PFK activity by phosphorylating the enzyme at the serine residues; and c) in order for cancer cells to meet their energy requirements due to rapid cell growth and division, the PFK1 enzyme is usually hyperactivated to cater for swift energy supply (13, 14). During rapid division in the cancer context, angiogenesis is always lagging, combined with even more slow maturation of the vessels. Such drastic changes in tumor mass induce hypoxia, which in turn leads to O-GlcNAcylation at serine 529 of PFK, giving cancer cells a selective growth advantage.
Given the critical role of PFK in glycolysis and cancer, and the lack of reports on PFK in bladder cancer, we conducted an in silico and in vitro study in an effort to depict the role of PFK family members in bladder cancer.
Materials and methods
Analysis of Public Dataset
The Cancer Genome Atlas (TCGA) database stores genomic and clinical data of a series of common cancers including bladder cancer; all data have been made public for analysis (15). The official TCGA report on bladder cancer was released and the database was analyzed on the cBioPortal platform (http://www.cbioportal.org/) (16). We used the provisional dataset for bladder cancer, which contained 126 MIBC samples whose somatic mutations, putative copy-number alterations, RNA-seq, and protein/phosphoprotein data detected with reverse-phase protein array (RPPA) were complete (17, 18). We input genes from the PFK family (PFKL, PFKM, PFKP, PFKFB1, PFKFB2, PFKFB3, and PFKFB4) and the cBioPortal automatically calculated the mutual exclusivity, frequencies of mutation and copy number variance (CNV), correlation between mRNA expression and CNV, coexpression analysis, enrichment analysis, survival data, and network analysis.
Cell Culture and Proliferation Assays
T24 and 5637 bladder cancer cells of urothelial origin were acquired from the cell bank of the Chinese Academy of Science (Shanghai, China). Cells were cultured with DMEM medium (Gibco) and 10% fetal bovine serum (Gibco). The PFK inhibitor 2,5-anhydro-D-glucitol-6-phosphate at 5 mM was used in all treatment groups. Cells were cultured for 72 hours and then processed with formalin fixation. After several rinses, cells were dyed using crystal violet, which was later dissolved by means of methanol. Proliferation was profiled by absorption at 540 nm read on the plate reader.
Measurement of Lactate Secretion
We followed a previously described protocol to measure the intracellular glycogen (19). In general, metabolite levels in the medium were measured using the lactate measurement assay kit (Sigma) as previously described.
Migration and Invasion Assay
Invasion and migration assays were performed as per established protocol (20, 21). In general, 5,000 cells were seeded in the top chamber of the Transwell upper chamber inserts that were either coated or uncoated with Matrigel for migration and invasion assays, respectively. The lower chambers were filled with complete media. Treated or untreated cells that transited through the membrane were stained with crystal violet and were counted under the microscope for 3 high-power fields.
Colony Formation Assay
The established protocol was reported previously (20, 22, 23). Roughly 1,000 cells were resuspended in complete medium mixed with 0.4% agarose. The mixture was layered on top of the solidified mixture of complete medium and 0.6% agarose. On top of both layers was 1 mL of complete medium. After 2 weeks of culture, plates were stained with 0.005% crystal violet for 1 hour. Colony tubercles were counted.
Statistical Analysis
In silico statistical analysis of the TCGA data was automatically performed through the cBioPortal web interface. All in vitro assays were run in triplicate in 3 independent experiments. Student's t-test was used to evaluate means between 2 groups. A p value of <0.05 was taken as statistically significant.
Results
PFK family was upregulated in bladder cancer
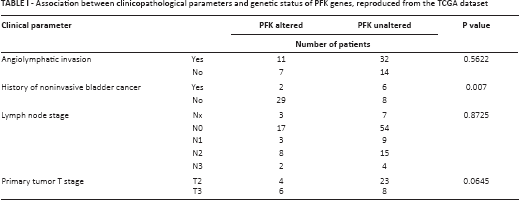
Using the TCGA database, we found that genetic alterations of PFK family members were prevalent in bladder cancer. In general, alteration of any PFK member occurred in 55 of 126 patients (~44%) (Fig. 1A). Gene amplification was the main type of alteration. Almost all alterations were towards enhanced gene function. Of note, alterations of PFKP and PFKFB3 were present in 13% and 14% of cases, respectively. We then studied whether amplification of the gene led to upregulation of mRNA and found a general trend of enhanced transcriptional activity in genetically amplified cases. The expression of PFKL and PFKP was consistently and gradually upregulated from gene deletion to diploidy, and eventually gene amplification (Fig. 1B-C). Nonetheless, in other genes of the PFK family, although a trend was observed, the upregulation of mRNA was not consistent with more gains in copy numbers (Fig. 2A-E), indicating that those genes exerted minor functions in bladder cancer. We also investigated the association between clinicopathological parameters and PFK gene status and found significantly more patients with a history of noninvasive bladder cancer harboring PFK gene alterations (p = 0.007; Tab. I). To sum up, almost half of the patients in the TCGA cohort had tumors with functionally enhanced PFK family genes, suggesting a role of PFK and glycolysis in bladder cancer.
Association between clinicopathological parameters and genetic status of PFK genes, reproduced from the TCGA dataset
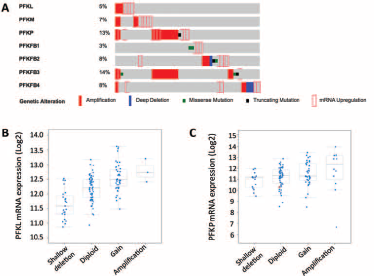
PFK members were generally upregulated in bladder cancer. Reproduced from
the TCGA bladder cancer database using the cBioPortal platform.
(
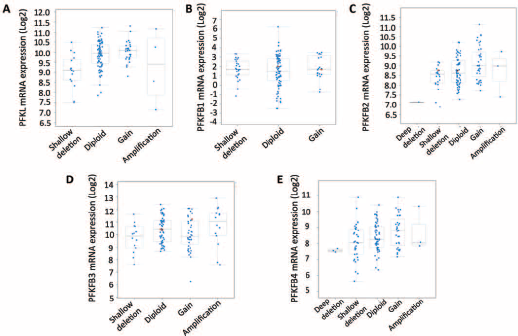
Expression levels of some PFK genes were discordant with copy number
variance. Reproduced from the TCGA bladder cancer database. The plot
function of the cBioPortal automatically calculated the mRNA expression
of PFKM (
Association between PFK Alteration and Survival
As the TCGA database included survival and recurrence data, we studied whether alteration of the PFK family as a whole had an impact on disease course. We found that the median survival time was 20.27 months in PFK-altered cases and 46.75 months in PFK-unaltered cases. However, the difference in survival was not statistically significant (Fig. 3A). Similarly, in the analysis of disease-free duration, we found that the median time to recurrence was 18 months in PFK-altered cases and 25.99 months in PFK-unaltered cases. The difference in time to recurrence was not statistically significant either (Fig. 4B). We concluded that PFK enhancement played a trivial role in the prognosis and recurrence courses of bladder cancer. The lack of statistical significance could be rooted in the nature of sophisticated networks in cancer development, in which a PFK-mediated metabolic shift contributed solely in part.
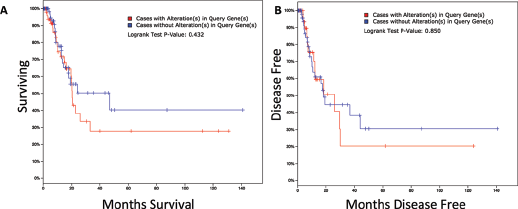
Alteration of PFK family genes was not associated with prognosis and
earlier recurrence. Reproduced from TCGA bladder cancer database. The
Kaplan-Meier curves were generated to demonstrate the overall survival
(
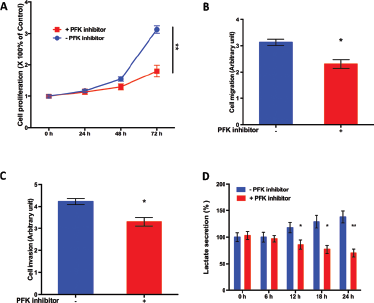
Inhibition of PFK reduced the growth of T24 bladder cancer cells.
(
Inhibition of PFK Attenuated Bladder Cancer Growth
As PFK gene products exerted critical functions in glycolysis (the Warburg effect), we speculated that inhibition of PFK function could attenuate glycolysis in bladder cancer and suppress cancer growth. By using the PFK inhibitor (2,5-anhydro-D-glucitol-6-phosphate), we found that inhibition of PFK significantly inhibited cell proliferation in T24 bladder cancer cells on day 3 of treatment (Fig. 4A). Of note, both migration (Fig. 4B) and invasion (Fig. 4C) were significantly stalled at 72 hours of treatment with the PFK inhibitor in T24 cells. In order to further explore the relevant mechanism, we measured the lactate level in the medium of T24 bladder cancer treated or untreated with PFK inhibitor. We found that PFK inhibition induced decreased lactate production compared both with the baseline level and untreated control over time (Fig. 4D). The timing (24 hours) of the difference in glycolysis levels following PFK inhibition was in accordance with the diversion in proliferation curves (Fig. 4A). All assays were repeated in 5,637 bladder cancer cells, where significant inhibition of cell proliferation, migration and invasion, and decreased lactate secretion were also noted (data not shown). In combination with previous results, we showed that PFK enhancement played a role in bladder cancer growth and contributed to compromised survival and earlier recurrence. Inhibition of PFK function using a specific compound attenuated the growth of bladder cancer cells via suppression of glycolysis.
Enrichment Analysis of PFK Gene Family
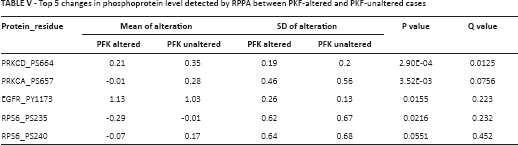
It has been reported that tumor cells enhance glycolysis not only to meet the need for rapid energy procurement but also to provoke a series of “by-product” pathways that contribute to tumorigenesis. By using the enrichment analysis function of cBioPortal, we were able to plot top ranking changes at the copy number, mutation, mRNA, and protein levels between cases with altered and unaltered PFK status. Significant changes at the mutational level are summarized in Table II. The co-occurrence and mutual exclusivity analysis indicated whether the mutated query gene tended to take part in or have redundant function with the PFK family. Significant changes at the copy number level are summarized in Table III. Significant changes at the mRNA level are summarized in Table IV. The mRNA level was plotted from the RNA-seq platform of the TCGA database. Finally, significant changes at the protein level are summarized in Table V. This table summarizes changes in phosphorylated proteins detected using the RPPA panel. Altogether, we observed a trend towards an increment in oncogenes and oncoproteins and suppression of tumor suppressor gene function.
Top 5 changes in gene mutation frequency between PKF-altered and PKF-unaltered cases
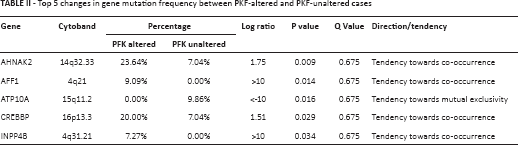
Top 5 changes in copy number variance between PKF-altered and PKF-unaltered cases
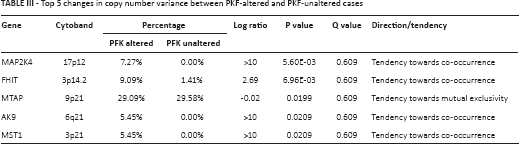
Top 5 changes in mRNA level detected by RNA-seq between PKF-altered and PKF-unaltered cases
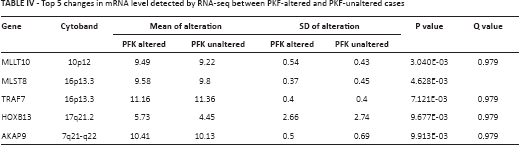
Top 5 changes in phosphoprotein level detected by RPPA between PKF-altered and PKF-unaltered cases
Discussion
Our study has been the first to address the role of PFK family genes in bladder cancer and has offered a contour of how this gene set could contribute to cancer development. We found that PFKP was not only amplified in a substantial group of bladder cancer patients but also hyperfunctional, along with a gain in copy number. These results, together with genetic alterations of other PFK members, indicated that not all PFK members performed evenly in bladder cancer. We therefore speculate that PFKP could exert a major effect in bladder cancer. Mammalian PFK1 is a 340 kD tetramer consisting of 3 subunits according to the tissue context, namely muscle (M), liver (L), and platelets (P) (24). In normal human cells, the PFK1 tetramer is tissue specific, i.e., the M subunit is only present in muscle tissue, whereas the L subunit is predominantly expressed in the liver and kidneys. Erythrocytes, on the other hand, can tetramize multiple forms of the enzyme as they express both L and M subunits. Changes in the PFK activity contribute substantially to the glucose metabolism in different tissues. It is thus intriguing that in our study all 3 isoforms were hyperactive in bladder cancer. We speculate that bladder cancer is in need of glycolysis to maintain its malignant potential. Also, the TCGA database included only MIBC, an aggressive subtype of bladder cancer. The increased glycolysis could contribute to its ability to evade and metastasize (25). As expected, our in vitro study showed that inhibiting PFK could combat against the cell motility of bladder cancer. We also found in the current study that patients with altered PFK gene status tended to have a history of noninvasive bladder cancer. Such a population is believed to have undergone several resections of the tumor before cystectomy. Therefore, one interpretation of the results is that PFK gene alterations could be more associated with a progressive phenotype and less with a phenotype that is initially aggressive.
PKF1 plays a key role in the glycolytic pathway as it curbs the step that is in charge of both energy release and the first irreversible glycolytic reaction (26). Such a metabolic switch mediates the precise control of the downstream elements of glycolysis. By contrast, glucose-6-phosphate can also join other elements of the glucose metabolism like the pentose phosphate pathway or glycogenesis upstream of the PFK1 switch. Activation of PFK1 activity is dependent in part on the ATP/AMP (adenosine monophosphate) ratio, as a high ATP level inhibits PFK1. This accounts for the initiation of glycolysis in case of energy shortage. In the current study, we have used 2,5-anhydro-D-glucitol-6-phosphate as the inhibitor of PFK activity. The compound exhibited potent inhibition of PFK in our in vitro assays. However, clinical application of the inhibitor still faces tremendous challenges and requires accurate validation, as broad inhibition of PFK could lead to serious adverse events.
Metabolic reprogramming is one of the characteristics of cancer. In particular, the major alterations in cancer cell metabolism are not focused on maximizing ATP production but on providing the ingredients needed to support macromolecular synthesis. In renal cell carcinoma, HIF1A increases the glucose uptake and controls the glucose flux through the glycolytic and pentose phosphate pathways (27). In our study, network analysis also designated HIF1A and MYC at the hub of the cross talk of PFK, both of which were key regulators between metabolism and multiple oncogenic pathways (Fig. 5).
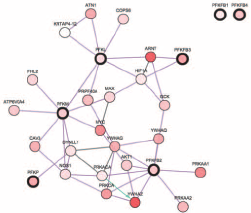
Reproduced from the TCGA bladder cancer database using the cBioPortal platform. The Network function demonstrated cross talk between PFK family genes.
In conclusion, our findings provide evidence that PFK family genes exert important functions in bladder cancer. Because novel treatment modalities for bladder cancer are urgently needed, targeting glycolysis via PFK could be a promising approach. However, the current targeted therapies aiming at metabolic shifts in cancer face an important challenge as resistance develops quickly in response to energy depletion. Our findings may serve to suggest a supplementary approach to the standard treatment modalities.
Footnotes
Financial support: None.
Conflict of interest: The authors declare there is no conflict of interest.