
Abstract
Annexin A1 is a 37 kDa calcium and phospholipid-binding protein that participates in several biological processes, such as inflammatory reactions, modulation of cell proliferation, regulation of cell death signaling, apoptosis, and, most importantly, tumor formation and development. Although annexin A1 has been implicated in the biology of various tumors, the findings are highly controversial and information regarding the underlying mechanism remains limited. Moreover, the mechanism by which annexin A1 participates in carcinogenesis and tumor progression is rather unclear. In the current study, we review the important biological functions of annexin A1 in different tumors. This work indicates that annexin A1 is a possible target for novel therapeutic intervention and that it is a potential biomarker for tumor diagnosis and screening.
Introduction
Annexin A1 (gene name, ANXA1) is the first member of the annexin superfamily to be characterized; it was originally known as a mediator of glucocorticoid-regulated inflammatory responses. In previous studies it was reported as macrocortin, lipomodulin, renocortin, and lipocortin-1, before it was named annexin A1 (1). The annexin superfamily is composed of 13 proteins that exhibit calcium-dependent binding to phospholipids. Annexin A1 has demonstrated complex roles in many diverse cellular functions, such as membrane interactions, inflammation, phagocytosis, regulation of proliferation, and cell apoptosis (1). Annexin A1 is also involved in carcinogenesis and/or tumor progression by participating in cell proliferation and differentiation, cell signaling, and metastasis (2). Although annexin A1 has been detected in a variety of tumors, the reported level of annexin A1 expression is inconsistent. Annexin A1 has been shown to be upregulated in some tumors but downregulated in others. Considering these contrasting findings, the mechanism by which annexin A1 participates in carcinogenesis and tumor progression remains unclear. Based on published reports and our research, we summarize these results and attempt to elucidate the possible mechanisms and intrinsic relationships of annexin A1 in cancer.
Molecular Structure of Annexin A1
The annexin superfamily shares a core that represents 80% of the protein and consists of 4 homologous repeated domains, approximately 70 amino acids long, that harbor ‘type 2’ calcium-binding domains. Each member of the superfamily is distinguished by a unique N-terminal region, which is related to the specific function and biological behavior of each member of the annexin superfamily (3). Annexin A1 is a 37 kDa protein consisting of 346 amino acids. Its N-terminal domain contains several putative Ser and Thr phosphorylation sites, as well as consensus sequences for glycosylation (Asn43–Ser45) and transglutamination. The N-terminal domain is embedded within the pore at low Ca2+ concentrations (4, 5) (Fig. 1). Previous reports show that annexin A1 contains the typical annexin core arranged around a pore, and has the ability to alter its conformation upon binding to calcium cations. The gene structure and sequence of annexin A1 have been successfully characterized in humans. Although the gene is multiexonic, spliced variants have not been presented so far. Multiple potential sites for the secondary processing of the protein have been found through sequence analysis. These sites can undergo acetylation and lipidation, as well as tyrosine, serine, and threonine phosphorylation, which all indicate that the protein may have multiple forms (6). Most importantly, in the case of Ca2+-free annexin A1 the N-terminal domain of annexin A1 is covered within the core of the protein. Upon binding to Ca2+, the N-terminal domain of annexin A1 is exposed and may participate in diverse biological functions through interactions with different molecules. Calcium ions mediate membrane binding and operate as an independent conformational switch that results in the emergence of the N-terminal domain (7) (Fig. 2).
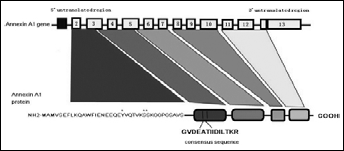
Schematic illustration of the localization, structure, and signaling components of the annexin A1 gene and protein. The N-terminal domain includes a 17-amino acid consensus sequence that is crucial to Ca2+ binding.
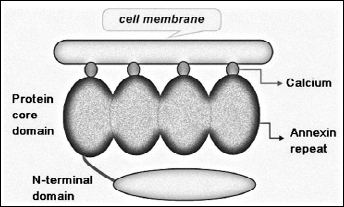
Structure of the human annexin A1 protein. Crystallography studies indicate that 4 repeated sequences are arranged around a pore and each of the 4 repeats in the core domain includes 17-amino acid consensus sequence. The N-terminal domain is embedded within the pore at low Ca2+ concentrations. Binding of annexin A1 to Ca2+ exposes its N-terminal domain, allowing it to participate in diverse biological functions through interactions with different molecules.
Expression Pattern of Annexin A1 in Tumors
Annexin A1 may have important regulatory roles in tumor development and progression, but these findings are highly controversial, and the mechanism of its involvement in carcinogenesis and tumor progression is unclear. The downregulation of annexin A1 expression in certain cancers (8–28) and the upregulation in other cancer types support this involvement (19, 28–45). Table I summarizes the studies performed on clinical cancer cells and tissues with decreased annexin A1 expression. The loss of annexin A1 expression has been observed in breast, gastric, esophageal, prostate, bladder, head and neck, laryngeal, and oral cancer, and correlates with tumorigenesis and malignant tendency. Annexin A1 expression has been linked to advanced stages of the specific cancers, as well as metastatic tendency and degree of differentiation. Thus, it has been proposed as an effective differential marker for the detection of these cancers. By contrast, Table II summarizes the studies performed on clinical cancer cells and tissues with increased annexin A1 expression. Upregulation of annexin A1 expression has been observed in lung, breast, pancreas, cervical, bladder, renal, and pituitary cancer. It was also significantly associated with advanced stages of disease, especially those with unfavorable prognosis. However, increased annexin A1 expression (32, 33, 37) and decreased expression have also been demonstrated in some similar cancer types (8–10, 19). The conflicting reports on the expression of annexin A1 in breast and bladder cancers remain unexplained. The conflict is hypothesized to be related to hormone receptor status, even though a clear correlation has not been reported. Interestingly, many studies suggested that variations in annexin A1 expression may be hormonally regulated (especially in breast cancer and pituitary tumors). These findings may be closely related to the known functions of annexin A1 in the proliferation and apoptosis of tumor cells. Recent studies have shown that cancer cells downregulate antiproliferative and/or proapoptotic proteins to express an oncogenic phenotype (1). An increasing number of studies are associating annexin A1 with various tumors, but the mechanism by which it influences tumor initiation and/or progression is still undefined. The specificity and functional diversity of annexins determines their N-terminal domain. Each member of the annexin superfamily interacts with different ligands, which leads to a variety of biological effects; thus, annexin A1 can potentially be linked to malignancy via numerous mechanisms (46). Other explanations proposed for these discrepancies include differences in organ specificities, cell culture strains, and mechanisms of tumor development and progression, as well as variations in the antibodies used. The aforementioned studies identify annexin A1 as a promising candidate for diagnosing certain cancers, but contradictory findings in cultured cell lines and tissues require more direct experimental evidence to resolve these problems using more clinical specimens and more rational experimental designs. However, the conflicting results regarding annexin A1 expression indicate its close association with tumor development and progression. Furthermore, new techniques of molecular biology, such as transfection and interference of the annexin A1 gene, creation of annexin A1-null mutant mice, and new technologies for gene and protein research will help clarify these discrepancies.
Summary of Reports of a Decreased Expression of Annexin A1 in Human Solid Tumors and Correlation with Clinical Outcome
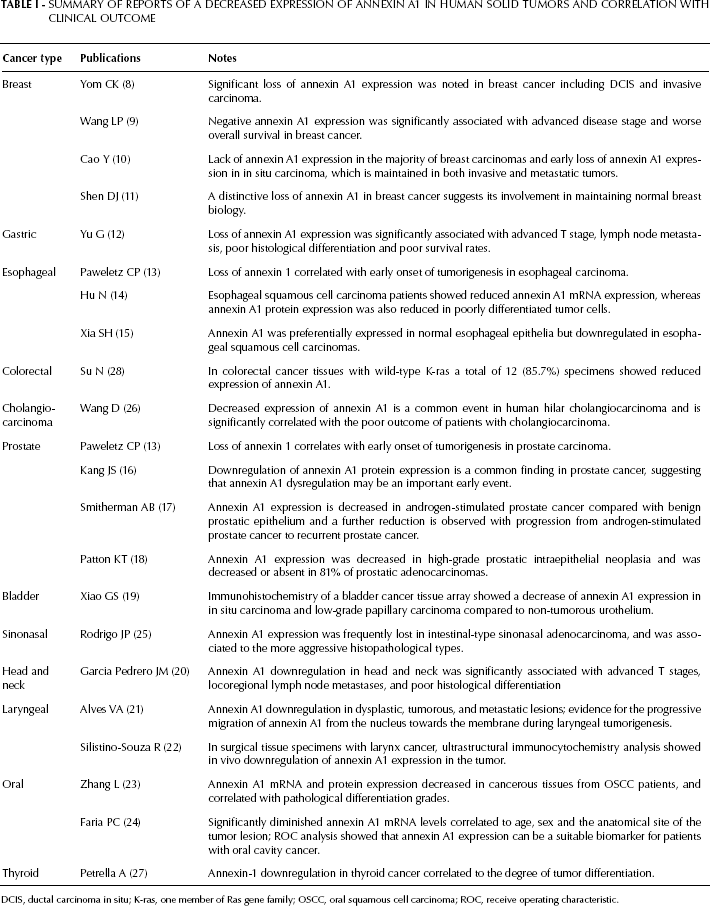
DCIS, ductal carcinoma in situ; K-ras, one member of Ras gene family; OSCC, oral squamous cell carcinoma; ROC, receive operating characteristic.
Summary of Reports of an Increased Expression of Annexin A1 in Human Solid Tumors and Correlation with Clinical Outcome
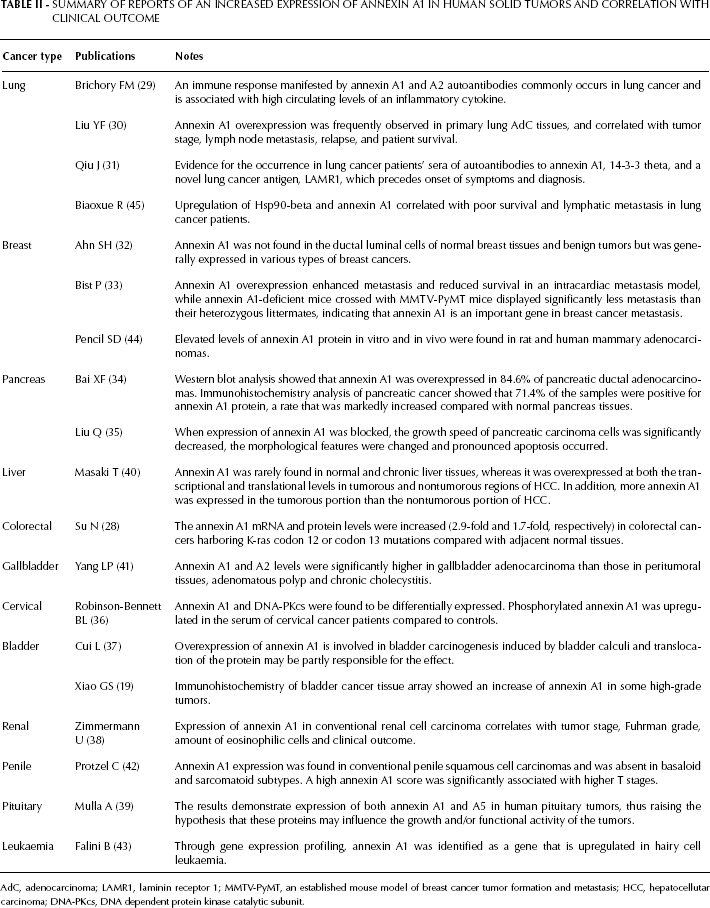
AdC, adenocarcinoma; LAMR1, laminin receptor 1; MMTV-PyMT, an established mouse model of breast cancer tumor formation and metastasis; HCC, hepatocellutar carcinoma; DNA-PKcs, DNA dependent protein kinase catalytic subunit.
Biological Function of Annexin A1 Involved in Cancer
Table III summarizes the existing research regarding the relationship between annexin A1 and cancer. Many reports suggest that annexin A1 is involved in tumorigenesis and cancer development. However, the exact mechanisms by which annexin A1 exerts its effects are still unclear. Numerous studies have reported the correlation between annexin A1 and tumor occurrence, aggressiveness, and metastasis. Gliomas overexpress the G protein-coupled chemoattractant receptor, formyl peptide receptor (FPR1), which promotes tumor progression when activated. Annexin A1 is present in tumor xenografts formed by human glioblastoma cells in nude mice; in addition, annexin A1 knockdown significantly reduces the tumorigenicity of glioblastoma cells in nude mice, and FPR1/annexin A1 double knockdown diminishes tumor growth even further (47). Annexin A1 is involved in constitutive nuclear factor-κB (NF-κB) activation, and suppression of annexin A1 in highly metastatic breast cancer cells impedes the migration and metastasis in vitro and in vivo (33). An in vitro invasion assay to evaluate the effect of siRNA-induced annexin A1 downregulation on the cell invasion capacity of A549 cells (human lung adenocarcinoma cell line) showed that introducing annexin A1 siRNA into A549 cells significantly decreases annexin A1 expression. The resulting annexin A1 downregulation decreases the in vitro invasiveness of the A549 cells (30). Previous studies have established a connection between tumor cell metastasis and epithelial-to-mesenchymal transition (EMT). One report showed that annexin A1 upregulation blocks EMT and ultimately affects breast cancer metastasis. Other studies have reported that increased annexin A1 expression decreases EMT in metastatic murine and human cell lines and in metastatic mouse and human carcinomas (48). RNAi-mediated annexin A1 knockdown induces TGFβ-independent EMT and metastasis in non-metastatic cells, which is caused by both annexin A1 downregulation and its downstream effect on the oncogenic Ras protein. Conversely, increased annexin A1 expression inhibits EMT and metastasis in metastatic murine and human mammary carcinoma cells. Therefore, annexin A1 potentially regulates EMT-like phenotypic switch (48). Some researchers hypothesized that the development and progression of invasive basal-like breast cancer involve the differential expression of annexin A1. Annexin A1 knockdown in invasive basal-like breast cancer cells restrains spontaneous lung metastasis, whereas annexin A1 overexpression enhances metastatic spread (49). Tumor initiation and progression are significantly increased in annexin A1-knockout mice, whereas these mice have longer survival times, with greatly increased tumor necrosis in laboratory animals. Vascular development and wound healing status have been measured to analyze the effects of annexin A1 knockout, and the results implied that angiogenesis and rebuilding are greatly retarded in knockout tissues. These results convincingly demonstrate that annexin A1 has pro-angiogenic functions in vascular endothelial cell sprouting, wound healing, and tumor growth and metastasis, thereby providing a new functional target for treating diseases such as cancer (50). In vivo experiments revealed that the expression of annexin A1 induces the growth and proliferation of pancreatic carcinoma cells. Conversely, transfecting annexin A1-siRNA into Suit-II cells (a pancreatic carcinoma cell line) significantly retards the growth and development of tumors. (51). To characterize annexin A1 involvement in dissemination of melanoma B16 cells (a melanoma cell line), investigators reduced the annexin A1 levels using siRNA and observed its effects in Matrigel-coated chambers. Interestingly, the metastatic progression of B16 cells decreased significantly, and several formyl peptide receptors were found by qRT-PCR, including FPR1, FPRrs1, and FPRrs2. Extensive research has shown that these receptors are involved in the invasion of melanoma cell lines. Incubating B16 cells with the FPR agonist fMLP significantly promotes the invasiveness of B16 cells in Matrigel-coated chambers, whereas incubating them with the FPR antagonist tBOC decreases their invasiveness. The results indicate that annexin A1 binds to FPRs, which leads to the invasion of melanoma cell lines and their subsequent metastasis (52). Knockdown of annexin A1 expression using siRNA significantly reduces tumor cell invasion. Translocation of annexin A1 from the cell interior to the cell surface occurs during cell migration, and functional inhibition of cell surface annexin A1 using antiserum significantly decreases the progression and metastasis of tumor cells. The presence of recombinant full-length annexin A1 promotes the invasion of SKCO-15, a colorectal adenocarcinoma cell line. In addition, a special peptide mimetic (Ac2–26) derived from the annexin A1 N-terminal also has the same effect (53). One study indicated that primary hepatocytes isolated from normal mice have low annexin A1 expression. Conversely, hepatocytes of antithrombin III SV40 T large antigen (ASV) transgenic mice express more annexin A1, and are more likely to develop into hepatocarcinoma. Furthermore, upregulated annexin A1 precedes the appearance of tumors, which demonstrates that the fluctuation of annexin A1 levels could help in diagnosing liver cancer (54). Other studies reported a correlation between annexin A1 and tumor cell apoptosis. Neutralization with anti-annexin A1 antibody or gene silencing with annexin A1 siRNA inhibits the apoptosis induced by FK228, a histone deacetylase inhibitor, which suggests that upregulated endogenous annexin A1 expression promotes cell death. FK228 induces high annexin A1 expression in Kasumi-1 cells (an acute myelogenous leukemia cell line), which promotes cell attachment and engulfment. Kasumi-1 cells are macrophages derived from human THP-1 (human acute monocytic leukemia cell line), and the abovementioned effect should be completely abrogated by annexin A1 knockdown via siRNA transfection or annexin A1 neutralization (55). Increased annexin A1 boosts cell viability, colony formation, and cell apoptosis. The epidermal growth factor (EGF) promotes cell proliferation and growth, but annexin A1 upregulation inhibits this process through other molecular ligands. Mitogen-activated protein (MAP) kinase family members p38 and JNK (c-JUN N-terminal protein kinase) are crucial molecules in cancer. One study indicated that restoration of annexin A1 expression promotes and induces the phosphorylation of p38 and JNK in prostate cancer cells (56). Another study observed that miRNA-196a is inversely correlated with annexin A1 mRNA levels in breast cancer cell lines, because miRNA-196a increases the tumor cell proliferation and independent growth, which suggest that it acts as an oncogene (57).
Functional Research of Annexin A1 in Cancer
FPR, formyl peptide receptor; NF-κB, nuclear factor kappa B; RNAi, RNA interference; TGFβ, transforming growth factor-β type; EMT, epithelial-to-mesenchymal transition; siRNA, small interfering RNA.
Annexin A1 Signal Pathway Involved in Cancer
Proliferation-Related Signaling Pathways
Annexin A1 has been linked with the reduced cell proliferation involved in the regulation of the ERK/MAPK signal transduction pathway. The ERK/MAPK pathway, which is involved in tumor development and progression, is activated by annexin A1 upregulation and leads to the reduction of tumor cell proliferation. This process is involved in the breakdown of the actin skeleton and downregulation of cyclin D1 expression (58, 59). The annexin A1 receptor FPR2 is mitogenic, and signaling through annexin A1/FPR2 increases the cyclin D1 levels, possibly through activation of the PI3K/Akt/p70S6K pathway (53, 60) (Fig. 3A). Annexin A1 expression levels are negatively correlated with interleukin 6 (IL-6) expression, which suggests that IL-6 enhances the initiation of prostate cancer and the progression of tumor cells. Decreased annexin A1 expression reportedly causes prostate carcinogenesis and promotes tumor aggressiveness by increasing IL-6 expression and activity (61) (Fig. 3B). Transcriptional activation of annexin A1 expression may be induced by the class I and class II histone deacetylase inhibitor, butyrate. The release of nuclear factor-Y (NF-Y) from the proximal CCAAT box has been suggested to mediate the effects of butyrate by enhancing p53 binding. The activity of p38 MAPK pathway induces the interaction of p53 with the promoter with or without butyrate. Further experiments showed that annexin A1 promoter activity and its protein expression require p38 MAPK activation by butyrate (62). Some researchers proposed that annexin A1 participates in cell proliferation as a substrate for the EGF receptor tyrosine kinase, thereby significantly inhibiting EGF-mediated proliferation (63). One study revealed that annexin A1 and alpha B-crystallin are important cellular proteins using two-dimensional proteomic and mass spectrometry analysis of metastatic MDA-MB-435 cells (human breast cell lines), but are downregulated in metastasis-suppressed BRMS1-transfected MDA-MB-435 cells. Furthermore, both of them mediated tumor metastasis suppression through BRMS1 (64) (Fig. 3C).
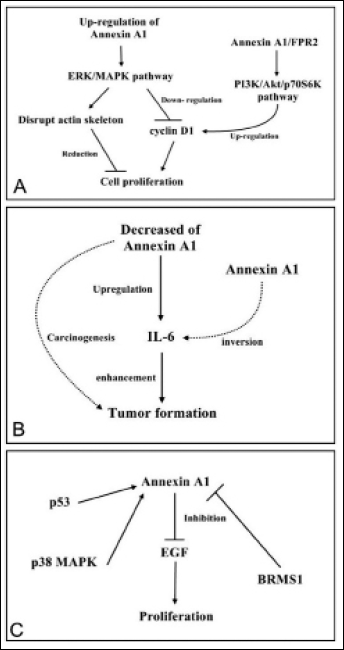
Signaling pathways involved in cancer cell proliferation. (A) Annexin A1 reduces cell proliferation by regulating the ERK/MAPK signal transduction pathway; (B) upregulated annexin A1 expression enhances tumor aggressiveness and proliferation via the upregulation of IL-6 expression and activity; (C) the interaction of p53 with annexin A1 is dependent on p38 MAPK activity. Annexin A1 is involved in cell proliferation as a substrate for the EGF receptor tyrosine kinase, and significantly inhibits the EGF-mediated proliferation. Annexin A1 mediates the suppression of tumor metastasis through BRMS1. ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; EGF, epidermal growth factor; BRMS1, breast cancer metastasis suppressor 1.
The aforementioned signaling pathways involve cancer cell proliferation. Upregulation of annexin A1 diminishes the proliferation of cancer cells, whereas downregulation of annexin A1 promotes cell proliferation.
Apoptosis-Related Signaling Pathways
The histone deacetylase inhibitor FR235222 upregulates annexin A1 expression at the transcriptional level. Moreover, FR235222 induces cell apoptosis in prostate cancer via a caspase-dependent mechanism. Conversely, downregulation of annexin A1 expression reduces FR235222-induced apoptosis, which suggests that annexin A1 expression is correlated with the proapoptotic effects, and that it regulates apoptosis (65) (Fig. 4A). Annexin A1 has previously been shown to be involved in the activation of p38 and JNK and appears to adjust signal transduction during proliferation and apoptosis. Restoration of annexin A1 expression activates phosphorylated p38 and JNK, and induces apoptosis (56). Overexpression of human PNAS4 (hPNAS4), a recently identified pro-apoptosis gene, induces apoptosis and cell cycle arrest in the S phase in A549 human lung adenocarcinoma cells. A previous study pointed out that the results may have been due to hPNAS4-induced apoptosis through annexin A1, and that the biological function of hPNAS4 must be investigated further (66) (Fig. 4B). Tumor necrosis factor (TNF)-induced apoptosis may involve activation of cytosolic phospholipase A2 (cPLA2), and annexin A1 was considered as an endogenous cPLA2 inhibitor. Dexamethasone increases annexin A1 synthesis and increases the resistance of U937 leukemic cells to TNF-induced apoptosis (67) (Fig. 4C). The aforementioned signaling pathways involve the apoptosis of cancer cells. According to previous studies, annexin A1 upregulation is correlated with the apoptosis of cancer cells through different mechanisms.
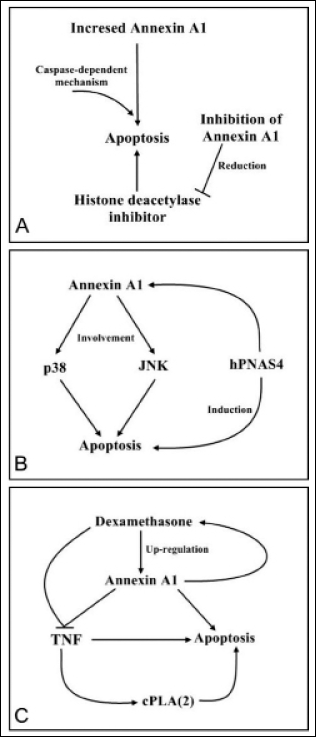
Annexin A1 signaling pathways involved in cancer cell apoptosis. (A) Histone deacetylase inhibitors increase the annexin A1 protein levels at the transcriptional level and induce apoptosis through a caspase-dependent mechanism. (B) The pro-apoptotic effect of annexin A1 involves the activation of p38 and JNK and the induction of hPNAS4, which appears to shift the balance of signal transduction away from proliferation and toward apoptosis. (C) Dexamethasone confers resistance to TNF-induced apoptosis in U937 leukemic cells (also through cPLA2 activation) by upregulating the intracellular annexin A1 levels and by facilitating a negative-feedback loop, which is activated upon stimulation with TNF. JNK, c-Jun N-terminal kinases; PNAS4, pro-apoptosis gene; TNF, tumor necrosis factor; PLA2, phospholipase A2.
Metastasis-related signaling pathways
Annexin A1 knockdown helps oncogenic Ras to induce TGFβ-independent EMT transition and metastasis in non-metastatic cells, whereas induced annexin A1 overexpression reverses EMT and abolishes metastasis (48). More recently, annexin A1 has been implicated in promoting metastasis formation by enhancing TGFβ/Smad signaling and actin reorganization. Annexin A1 facilitates an EMT-like switch and enhances the migration and invasion of metastatic breast cancer cells (49) (Fig. 5A). Annexin A1 expression is correlated with NF-κB activity in metastatic breast cancer (68), which indicates that annexin A1 is involved in the constitutive activity of IκB kinase (IKK) and NF-κB. CXCR4 and uPA, 2 important downstream targets of NF-κB, are modulated by annexin A1 silencing. CXCR4 induces the migration of breast cancer cell lines, and CXCL12 is required in tissue-specific migration; however, more importantly, tissue-specific migration modulates the combined effect of CXCR4 and CXCL12, which suggests that annexin A1 is crucial in the process of metastasis of breast cancer cell lines (33) (Fig. 5B). VEGF induces annexin A1 phosphorylation, whereas inhibiting p38 impairs annexin A1 phosphorylation. VEGF-mediated activation of LIM kinase 1 downstream of the p38 pathway induces annexin 1 phosphorylation. Furthermore, siRNA-mediated knockdown of annexin A1 blocks VEGF-induced cell migration, which is conversely enhanced by annexin A1 upregulation (50, 69). Annexin A1 is phosphorylated by a number of kinases, including EGF-R tyrosine kinase, PKC, PDGFR-TK, and HGFR-TK to mediate proliferation (1). Annexin A1 is also crucial in liver regeneration and tumorigenesis. Several important molecules are modulated by annexin A1 including cPLA2, EGF-R, and EGF, and they function during liver tumorigenesis via related signal pathways (54, 70) (Fig. 5C). Figure 5 shows that annexin A1 is involved in the migration of cancer cells and that its phosphorylation induced by different kinases and growth factor receptors affects the migration of cancer cells.
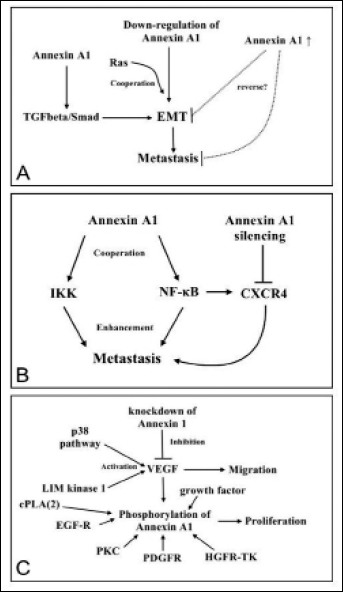
Annexin A1 signaling pathways involved in the migration of cancer cells. (A) Annexin A1 downregulation cooperates with oncogenic Ras to induce TGFβ-independent EMT and metastasis; (B) Annexin A1 expression is correlated with NF-κB activity, and is required for the constitutive activity of IκB kinase (IKK) and NF-κB in highly metastatic breast cancer. Downstream targets of NF-κB include uPA and CXCR4, which are modulated by annexin A1 silencing. (C) Annexin 1 regulates the angiogenic effect via VEGF, and this process depends on the activation of the p38/LIM kinase 1. Annexin A1 is phosphorylated by EGF-R tyrosine kinase, PKC, PDGFR-TK, HGFR-TK, cPLA2 activation, or EGF-R and EGF function to mediate proliferation. Ras, ras gene; TGFβ, transforming growth factor-β; EMT, epithelial-to-mesenchymal transition; NF-κB, nuclear factor kappa B; uPA, urokinase-type plasminogen activator; CXCR4, chemokine (C-X-C motif) receptor 4; VEGF, vascular endothelial growth factor; EGF-R, epidermal growth factor receptor; PKC, protein kinase C; PDGFR-TK, platelet-derived growth factor receptor tyrosine kinase; HGFR-TK, hepatocyte growth factor receptor tyrosine kinase; cPLA2, phospholipase A2.
Drug Resistance Modulation of Annexin A1 in Cancer
The use of anti-cancer chemotherapy often promotes multidrug-resistant (MDR) tumors. The loss of annexin A1 may improve the resistance of cancer cells to apoptosis induced by chemotherapeutic agents. Thus, annexin A1 is a potential negative biomarker in cancer treatment assessment, and will be valuable in further studies on the mechanisms of MDR. Previous studies reported the over-expression of annexin A1 in several MDR tumor cell lines; additionally, total RNA analysis from drug-sensitive and drug-resistant cells revealed an increase in annexin A1 mRNA in drug-resistant cells relative to drug-sensitive cells. Transfection of annexin A1 cDNA into drug-sensitive MCF-7 cells (human breast cancer cells), results in a twofold to fivefold increase in the resistance of transfected cells to adriamycin (ADR), melphalan, and etoposide. By contrast, transfection of reverse annexin A1 cDNA into SKOV-3 cells (ovarian cancer cells) decreases the expression of annexin A1 and causes a threefold to eightfold increase in sensitivity to these drugs (71). Cytotoxic activity and drug resistance effects related to dexamethasone were discovered in PC-3 cells (androgen-independent prostate cancer cells), and this action can be mediated by annexin A1 induction. These results indicate that antagonistic antitumor effects may be stimulated by combining dexamethasone with conventional chemotherapeutic agents (72). The effect of chemotherapy is often hindered by MDR in the treatment of chromic myeloid leukemia (CML). To unravel the factors related to MDR and to understand its possible mechanisms, annexin A1 was downregulated in the ADR-resistant human erythroleukemia cell line K562/ADR. Further analysis showed that decreased annexin A1 expression plays a prominent role in the observed drug resistance in K562/ADR cells. These data have fundamental significance for further research regarding the mechanisms of MDR, and may also indicate a potential new diagnostic marker for the effect of chemotherapy (73).
Problems and Prospects
Published reports implicate annexin A1 in a broad range of molecular and cellular physiological processes. Annexin A1 is a mediator of the antinflammatory activity of glucocorticoids in the host defense system (1). It also contributes to the regulation of a variety of inflammatory pathways and other cellular processes, such as cell proliferation, cell death signaling, differentiation, and apoptosis, especially in various types of cancer. However, while annexin A1 was found to be differentially expressed in various tumors, the findings on this topic are highly controversial, and the underlying mechanism remains unknown. Moreover, the mechanism by which annexin A1 participates in carcinogenesis and tumor progression is unclear because of the controversial results about annexin A1 in cancer. Based on published reports and our recent work, we considered that alterations of annexin A1 expression may be associated with several tumor characteristics: (i) the differential expression of annexin A1 in various tumors may be caused by differences in histological origin and tumor stage. More research work shows that annexin A1 expression in squamous cell carcinoma is higher than in adenocarcinoma, but the underlying mechanism remains unknown. (ii) The high annexin A1 expression in normal tissues appears to be reduced during tumor transformation, but low annexin A1 expression in normal tissues increase during tumor transformation. Annexin A1 is potentially linked to malignancy via numerous mechanisms because annexins bind to many different ligands. Although this phenomenon has been observed in several studies, the underlying mechanism is still unclear. (iii) The discrepancies in annexin A1 expression may be associated with different research environments relating to organ specificity, alterations during cell culture, dynamic changes in expression during tumor progression, and/or variance in the antibody reagents used. Current opinion considers that annexin A1 is a calcium-binding protein with a conserved domain (annexin domain) that binds to phospholipids. The conserved terminal domain may determine expression specificity and provide functional diversity (2). Differences in biological functions and signal pathways involving annexin A1 have also been reported in different tumors. These discrepancies are attributed to the differential expression patterns and the complex biological behavior of annexin A1. Moreover, a number of studies have shown that annexin A1 is correlated with apoptosis regulation; some groups suggested that annexin A1 is a pro-apoptotic factor, whereas others suggested that annexin A1 is involved in apoptosis resistance. The actual mechanism behind this apparent contradiction is unclear, but could depend on the different cell types or different cellular phenotypical variations (2).
Conclusion
Considering the complex correlations between annexin A1 and cancer, alterations in annexin A1 expression in cancer cells may affect different patterns of malignant cellular behavior, such as metastasis, tumor cell apoptosis, and tumor cell proliferation, depending on its functional diversity. Numerous research findings indicate that annexin A1 may be a potential therapeutic target in the treatment of malignant disease. Moreover, the aforementioned studies also indicate that annexin A1 is a promising tumor biomarker. However, the precise functions of annexin A1 in different cancers are still poorly understood. Some contradictory findings in different tumor cells involving different biological conditions and distinct cultured cell lines need to be clarified through direct analysis of large samples of primary tumor tissue. Clarification of these contradictory lines of evidence will require analysis of annexin A1 gene knockdown and transfection using advanced biological methods. The in-depth study of annexin A1 features will help understanding the mechanism of tumor invasion and tumor metastasis, and help determining if annexin A1 can be an indicator for tumor invasion, metastasis, and prognosis. This will also open up new paths for the prevention and treatment of malignant tumors.
Footnotes
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities. The authors wish to thank Drs. WU Dian-lei and Wang Xin-hong for their helpful discussions, critical comments provided during the preparation of this manuscript and also wish to thank all authors of references.