
Abstract
Background
The amorphous poly(lactic acid-co-lysine) (PLL) with hydrophilic reactive groups was synthesized by the condensing copolymerization and the blending miscibility of poly(L-lactic acid) (PLLA) and PLL was investigated.
Methods
The miscibility of PLLA and PLL was evaluated by analyzing the thermal properties, crystallization behaviors, crystal morphologies and mechanical properties of the PLLA/PLL blends.
Results
The results indicated that amorphous PLL changed the crystallization behaviors and thermal properties of PLLA, decreasing the crystallinity of PLLA in the blends. The amorphous PLL molecule chains in PLLA/PLL blends were able to enter into the lamellars of PLLA spherulites and affect the crystallization behaviors of PLLA, resulting in imperfect, spherulite structures of PLLA. The formation of hydrogen bonds between PLLA molecular chains and PLL molecular chains enabled partial compatibility in this blend system.
Conclusions
The existence of PLL improved the hydrophility of PLLA/PLL blends, led to a higher content of PLL in the PLLA/PLL blend system, and better hydrophilic properties of the blend system. The PLL was partially miscible with the PLLA, and the PLLA/PLL blend with 10 wt% PLL had improved tensile properties.
Introduction
Owing to good biocompatibility and biodegradability, poly(lactic acid) (PLA) has wide applications in clinical medicine, such as surgical sutures (1), antibacterial materials (2), drug delivery systems (3), tissue engineering scaffolds (4), and so on. However, poly(L-lactic acid) (PLLA) has high crystallinity, poor impact toughness and a long degradation period, so its application has been severely restricted. To satisfy the requirements for new, high-value applications, the specific properties of PLLA need to be improved, including hydrophility, high impact toughness, proper degradation cycle, and thermal stability. These properties of PLLA can be modified by plasticization (5), copolymerization (1, 6), blending (7), molecular modification (8), and composite modification (9), etc., among which blending is an efficient method expected to change the morphology and improve the properties of PLLA. Studies about PLLA blending with nondegradable polymers (poly(vinylidene fluoride) (10), poly(methyl methacrylate) (PMMA) (11), acrylonitrile-butadiene-styrene copolymer (ABS)(12), etc.) and semi-crystalline degradable polymers (poly(ε-caprolactone) (13, 14), poly (ethylene oxide) (15), etc.) have been studied sufficiently at present. However, blending with a nondegradable polymer would decrease the degradable properties of PLLA. Blending PLLA with the amorphous polymer could improve its performance in an evident fashion because of its particular morphologies and properties only if the PLLA has a certain miscibility with the amorphous polymer. Unfortunately, PLLA has low miscibility with most of the amorphous polymers – for example, the PLLA/ PMMA and PLLA/ poly(D, L-lactic acid) (PDLLA) blend (16, 17). The miscibility of the PLLA and the amorphous polymer could be improved if the amorphous polymer possesses segments of oligomer lactic acid or the functional groups that can form hydrogen bonds with the PLLA.
In this paper, L-lysine with good biocompatibility and multifunctional groups was chosen as the co-polymer monomer for L-lactic acid, and the poly(lactic acid-co-lysine) (PLL) were synthesized by the direct melt polycondensation via molecular design. The amorphous PLL, copolymers of L-lactic acid and lysine, possesses improved reactive functionality and hydrophility, and the amino acid groups on the PLL macromolecules with the carbonyl groups of the PLLA were able to form hydrogen bonds (18, 19), providing PLL with a strong intermolecular force with the PLLA. That is, the miscibility of PLL and PLLA were enhanced, allowing the properties of their blend to be improved. Subsequently, a blending system with different proportions of amorphous PLL and semi-crystalline PLLA was prepared by the solvent casting method. The blending miscibility between PLLA and PLL was accessed by investigating the thermal properties, crystallization behaviors, crystallization morphologies, mechanical properties and hydrophilic properties of the PLLA/PLL blends. The blends had the distinct crystallization behavior, reactive functionality and hydrophilicity from PLLA with the addition of PLL while retaining good biocompatibility.
Materials and Methods
Experimental materials
L-lactic acid solution was 80 wt% analytical reagent, purchased from Aladdin Industrial Corporation; L-lysine (99.5 wt%) was provided by Sigma-Aldrich; poly(L-lactic acid) was supplied by Shanghai Leon Chemical Ltd. with a molecular weight (Mw) of 305 × 103, number-average molecular weight (Mn) of 221 × 103, and polydispersity index (PDI) of 1.38; N, N-dimethyl formamide (DMF) and chloroform were purchased from Aladdin Industrial Corporation; phosphoric acid solution was purchased from Sigma-Aldrich with a concentration of 85 wt%.
Synthesis of PLL
The synthesis of PLL was carried out in nitrogen atmosphere. The molar ratio of L-lactic acid and L-lysine was 9:1, and the proportion of phosphoric acid in the mixture of L-lactic acid and L-lysine was 0.8 wt%. The L-lactic acid solution (8.109 g), the lysine solution (1.288 g) and the phosphoric acid solution (0.073 g) as the catalyst were added to a flask with stirring. The mixture was heated to 155oC for 12 hours. The byproduct water was removed by pumping. After the reactions were completed, the reaction products were cooled down and the DMF was added to dissolve the solids. After the dissolution process, the products were purified with deionized water by precipitation method. The purified solids were dried at 40oC in a vacuum oven, then the ultimate dark yellow PLL was obtained.
Preparation of PLLA/PLL blends
PLLA/PLL blends were prepared by the solvent casting method. First, the proportions of PLLA and PLL were 95: 5, 90: 10, 80: 20, 70: 30, 60: 40, 50: 50, 40: 60, 20: 80, respectively, and the mass of each proportion was 1.5 g. These solutions with concentrations of 6.5 wt% were prepared by adding the chloroform (14.4 mL) as solvent, then sealed and low-speed stirred for 12 hours. Last, these solutions were cast in flat Petri dishes and dried in a vacuum oven at room temperature for 24 hours (pressure <400 Pa). The casting sheets were cut to the required sizes and set aside. The prepared blends were named PLLA95/PLL5, PLLA90/PLL10, PLLA80/PLL20, PLLA70/PLL30, PLLA60/PLL40, PLLA50/PLL50, PLLA40/PLL60, and PLLA20/PLL80 in turn.
Characterization of PLL
The molecular chain groups and the structure of PLL were determined by Fourier transform infrared spectroscopy (FTIR; Thermo Scientific), an elemental analyzer (Elementar Trading) and nuclear magnetic resonance (NMR; Bruker BioSpin) (CDCl3 as solvent, 25oC). The molecular weights and molecular weight distributions of PLL were analyzed by gel permeation chromatography (GPC-PL50; Waters Corporation) with the chloroform as solvent at 25 oC, a solution concentration of 0.10 mg mL-1, a flow rate of 1.00 mL min-1, and column height of 650 mm. Glass transition temperature (Tg) of PLL were determined by a differential scanning calorimetry (DSC; Mettler Toledo).
Characterization of the PLLA/PLL blends
The thermal and crystal properties of PLLA/PLL blends and PLLA were analyzed by DSC and the glass transition temperature (Tg), the melting point (Tm), the fusion heat (ΔHf), and the crystallinity (Cr) were determined. Each sample of about 8 mg was heated from 0 to 200oC with the heating rate of 20oC min-1 under nitrogen first. After staying for 5 minutes, the blend was cooled to −20oC at the cooling rate of 10oC min-1. Subsequently, the sample was heated from −20oC to 200oC at the heating rate of 10oC min-1 after staying for 5 minutes at the end of cooling period. The crystallinitiy of the blend was calculated according to the melting heat of the DSC heating curve by Equation [1]. The crystallinity of PLLA in the blend was calculated by Equation [2].
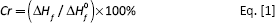
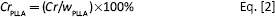
where ΔHf is the heat fusion of PLLA, ΔH0f is the melting heat of the complete crystalline PLLA and its value is −93.7 J g-1, wPLLA is the PLLA content (wt%) in the blend, and CrPLLA is the crystallinity of PLLA in the blend.
The crystalline morphology of PLLA/PLL blends could be observed by a polarizing optical microscope (POM; Nikon). The blends were heated to melt at the rate of 10oC min-1 to 210oC. After maintaining the temperature of 210oC for 5 minutes, the melting blends were pressed gently into thin slices to remove air under nitrogen, then cooled slowly to 120oC at the rate of 1oC min-1. During the cooling process, the temperature of the spherocrystal that were appearing, the growth rate of the spherulites, and the maximum size of the spherulites were observed and detected.
X-ray diffraction (XRD, Bruker AXS) were used to investigate the crystal structure of PLLA and PLLA90/PLL10 films. The tube voltage and current were 40 kV and 40 mA with the scanning speed 8oC min-1. The hydrophilic properties of PLLA/PLL blends were characterized by a contact angle measuring device (Shanghai Zhongchen Digital Technic Apparatus). PLLA/PLL blends and PLLA about 1 g were pressed into the films with the 1 mm thickness, and the contact angles of the films were detected. The tensile properties and fracture surface morphologies of PLLA/PLL blends were characterized. Slices of PLLA and PLLA/PLL blends with a height of 0.5 mm were prepared by a double-screw extruder (Lanzhou LAN Tai Plastic Machinery). The extruding temperatures of the feeding section, compression section and homogenization section were 140oC, 170oC, 180oC, respectively, and the die temperature was 175oC. The slices were cut into rectangular samples of length 60 mm and width 6.2 mm. The tensile strength and elongation at break were evaluated by an electronic tensile tester (Shandong Banner International Trade) with a stretching rate of 5 mm min-1. The tensile fracture surface morphologies of PLLA/PLL extrusion sheets were observed by SEM.
Results and discussion
Polymerization mechanism of PLL
When L-lactic acid and lysine are copolymerized, there occur some possible chemical reactions including self-polymerization of L-lactic acid (condensation of hydroxyl and carboxyl), self-polymerization of lysine (ω-amino and/or α-amino reacting with α-hydroxyl) and the condensation polymerization of lactic acid and lysine. The condensation reaction of lactic acid and lysine may occur and the possible reaction productions are shown in Figure 1, since there are 2 kinds of amino groups (ω-amino and α-amino) in the lysine molecule.
The reaction of Figure 1A is more likely to occur because ω-amino is more active than α-amino in lysine (Fig. 1B). The ω-amino is apt to react with the carboxyl group in lactic acid or carboxyl in lysine during the condensing polymerization.
Structure characterization of synthesized PLL
The structure of the synthesized PLL was verified by FT-IR, elements analysis, 1H-NMR and 13C-NMR, respectively, and the results are shown in Figures 2 to 4. Figure 2 is the FT-IR spectrum of PLL, where the stretching vibration of -NH- was at 3391 cm-1 and the stretching vibration of methyl was near 2993 cm-1. The peaks of 2943 cm-1 and 1452 cm-1 showed the stretching vibration and bending vibration of C-H, respectively. The stretching vibration peak of 1751 cm-1 meant carbonyl and the stretching vibration peak of 1663 cm-1 represented the amide group. The bending vibration peak of methyl was near 1380 cm-1 and the stretching vibration of C-O was at 1261 cm-1.
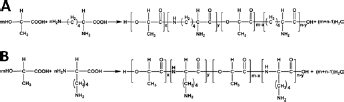
Possible copolymerization reactions of L-lactic acid and lysine.
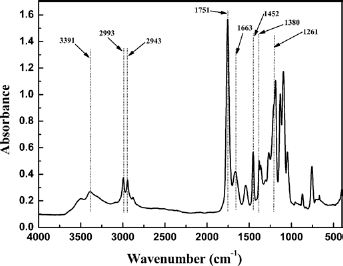
FT-IR spectrum of PLL.
The 1H-NMR spectrum of PLL is shown in Figure 3, in which δ 7.27 ppm indicates the H on -NH- and δ 5.16 ppm represents the H in -O-CH-CO-. δ 3.24 ppm shows the H of CH in -CH-NH2 and δ 2.94 ppm shows the H of CH2 in -NH-CH2-. δ 1.78 ppm (e), 1.48 ppm (g), 1.31 ppm (h) indicates the H of -CH2-CH2-CH2-, respectively. δ 1.59-1.47 ppm indicated the H of -CH3.
The lactic acid units contained the methyl and methenyl groups, while the lysine units had the methylene and methenyl groups. The integration of the proton peak is also shown in Figure 3. Due to the overlap of the methyl and methylene peaks (Fig. 3e–h), the proportion of each repeating unit in PLL could be obtained by calculating the peak areas of the methylene in -NH-CH2- and the methenyl in the lactic acid units and the lysine units. The area ratio of b peak and d peak was 1:0.22, which proved the ratio of methenyl and -NH-CH2- group was 1:0.11. That is, the ratio of the lactic acid unit and the lysine unit in the PLL was 89:11, while the theoretical value of the ratio was 90:10 according to the monomer feed ratio. This indicated that the actual content of lysine units in the PLL was a little higher than the theoretical content.
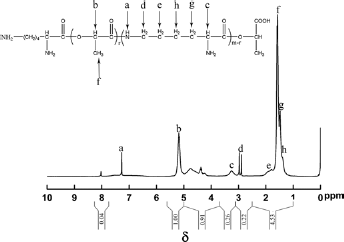
1H-NMR spectra of PLL.
In order to detect the content of the 2 monomers in the copolymer, the elemental analysis was applied to determine the quality percentage of the various element contents. The theoretical contents of various elements were calculated according to the reaction mechanism, thus the theoretical value and the tested value of lysine units content could be obtained. Table I is the result of elemental analysis.
The result of elemental analysis
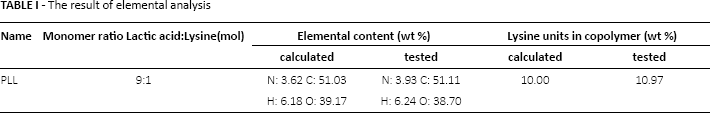
As shown in Table I, the tested value of each element content in the copolymer was close to the theoretical value, and the proportion of the 2 units was close to the monomer feed ratio, which indicated that the both monomers participated in the polymerization reaction, and the 2 monomers had a similar conversion rate. The tested nitrogen content in the copolymer was higher than the theoretical content, illustrating that the content of lysine units in the copolymer was a little higher than that of the lysine feed ratio. The main reason was that the L-lactic acid was liable to loss by evaporating under the temperature at the early stage of the copolymerization. This was different from the copolymerization of the poly(aspartic acid -co- lactic acid) (PAL) (20), where the ratio of aspartic acid units in the PAL was much higher than that of the aspartic acid feed ratio, because of the sublimation of the L-lactide at high reaction temperature (180-210oC). In the synthesis of PLL, the L-lactic acid was used as the reactant and it evaporated a little at the reaction temperature 155oC, so the tested percentage of lysine units in the copolymer was close to the theoretical content. The result of elemental analysis was consistent with the results of 1H-NMR analysis.
Figure 4 shows that the 13C-NMR spectrum of PLL. δ 169.5 ppm evinced the C in -CO-O-. δ 163 ppm represented the C of -CO- and δ 77.4 ppm, 77.2 ppm, 77.0 ppm, 76.6 ppm indicated the C of -O-CH-. δ 71.6 ppm and 69.6 ppm represented the C of-CH2-CH2-CH2-CH2-, δ 66.6 ppm and 66.5 ppm represented the C of -CH2-NH2, and δ 20.3 ppm indicated the C of -CH3 in CH-CH3.
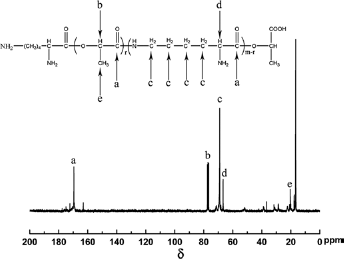
13C- NMR spectrum of PLL.
It can be confirmed from the FTIR, elemental analysis, 1H- NMR and 13C- NMR that the structure of the PLL contained amino groups, amide groups, ester groups, methyles, and methylenes, which showed that the structure of the product included both poly(lactic acid) segments (ester groups, methyls and methylenes) and polylysine segments (amino groups, amide groups, ester groups). The results indicated that the copolymer of poly(lactic acid-co-lysine) (PLL) was synthesized by the copolycondensation of lactic acid and lysine.
Molecular weight of synthesized PLL
The GPC curve of PLL is shown in Figure 5, showing that the poly(lactic acid-co-lysine) had the sole relative molecular mass peak, and the molecular weight distribution was an approximate normal distribution. The Mw was 19.8 × 103, Mn was 17.7 × 103, Z-average molecular weight (Mz) was 22.2 × 103, and the polydispersity index (PDI) was 1.12. The results illustrate that the product was a copolymer with a single relative molecular weight peak and narrow molecular weight distribution.
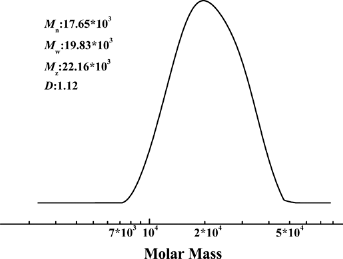
GPC curve of PLL.
Racemization of polylactic segment in PLL
High-resolution NMR was used to analyze the stereoregularity and racemization of the carbon chains of both homopolymers and copolymers (11). δ 169.5 ppm (carbonyl) and δ 69 ppm (methyne) in 13C-NMR spectrum and δ 5.18 ppm (methyne) in 1H-NMR spectrum were analyzed to determine the stereoregularity and racemization of the poly(lactic acid) segments in PLL, thus the mechanism of co-polymerization was explored. Figures 6 and 7 are the spectra of carbonyl 13C-NMR and the methyne 1H-NMR.
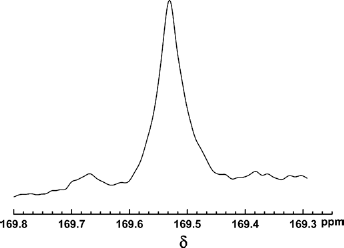
Carbonyl 13C-NMR of poly(lactic acid) segments in PLL.
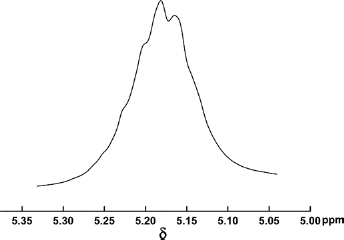
Methyne 1H-NMR of poly(lactic acid) segments in PLL.
According to the literature (21), PLLA molecular chains without racemization possessed high stereoregularity, in both their carbonyl and methyne there occurred single sharp peaks in the 13C-NMR spectra and this kind of PLLA molecular chain presented as semi-crystalline with high crystallinity. The typical quartet peaks of methyne in the 1H-NMR spectrum was also used to analyze the racemization of PLLA (22). From Figures 6 and 7, 13C-NMR detected that the split and migration of methyne and carbonyl peaks in the poly(lactic acid) segments of PLL occurred, and the methyne peak in 1H-NMR spectrum was composed of several nonconsistent peaks with significant deviation. The results showed that poly(lactic acid) segments in PLL had different spatial configurations, indicating that the ester exchange reaction and chain transfer reaction occurred in the copolymerization and caused significant racemization. PLL is likely an amorphous polymer due to the syndiotactic or atactic poly(lactic acid) segments in PLL.
Thermal performance analysis of PLLA/PLL blends
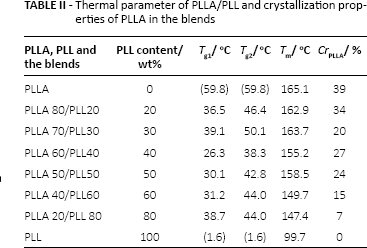
The Tg, Tm and melting heat of PLLA, PLL and PLLA/PLL blends of different proportions were detected by DSC, thereby allowing the crystallinity of PLLA in the blends to be calculated. Figure 8 shows the heating curves of different materials. Table II shows the thermal parameters of PLLA/PLL blends with different proportions and the crystallization properties of PLLA in the blends.
Thermal parameter of PLLA/PLL and crystallization properties of PLLA in the blends
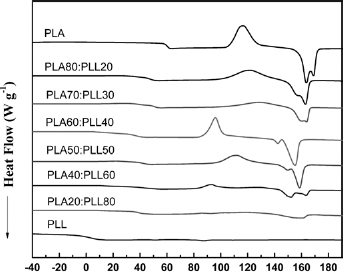
DSC heating curves of PLLA, PLLA/PLL blends and PLL.
The DSC curve of PLL showed that the melting peak of DSC was not significant. That is, PLL was an amorphous polymer. The Tg of PLL was 1.6oC. From Figure 8 and Table II, there were 2 Tgs in the DSC heating curve of each PLLA/PLL blend. If the 2 Tgs of the blend that were very close to those of the pure polymers, the 2 polyesters were confirmed to be immiscible in the blend, for example, in the PLLA/PCL blend (13). In this study, the Tg values were neither close to the pure polymers nor dependent on the composition strictly, which suggested that the blends were partially compatible. From Table II, the Tg2 of the blends was closer to the Tg of pure PLLA, but significantly decreased with the increase of PLLA content, which was particularly evident in PLLA60/PLL40 and PLLA50/PLL50. The results were similar to the addition of the plasticizers, which decreased the Tg of PLLA sharply (5, 23, 24). When PLL molecules entered into the PLLA chains, the intermolecular forces of PLLA molecular chains became weakened and the PLLA molecular chains became flexible; simultaneously the activities strengthened and the crystallinity decreased. The Tg1s of the blends were in between the Tg of PLL and that of PLLA owing to the compatible parts of the 2 portions.
For the completely immiscible system, the crystallinity of crystalline component was not affected by the blending component. For PLLA/PLL blends, the crystallinities of PLLA (CrPLLA) in the blends changed with the different components, which was different from the completely immiscible system (25, 26). The addition of amorphous PLL into the blends had an influence on the crystallinity behaviors of PLLA in the blends because some amino groups and terminal hydroxyl groups in PLL molecular chains were able to form hydrogen bonds with the carbonyl groups in PLLA, enhancing the interaction between 2 macromolecules and impeding the crystallization. The results proved that the PLLA was partially compatible with PLL.
The melting points of the blend system decreased with the increase of PLL contents. The reason for the melting point decrease was explained by a strong interaction between the 2 kinds of polymers in the melting state (27). That is, the addition of PLL resulted in the increase of the PLLA crystalline imperfections. In all, there were 2 reasons for the melting point decrease of the PLLA in the blends: one was the thermodynamic aspects, and the other was the morphological structure. In thermodynamics, the melting point decline of blends was considered as the performance of compatibility. Assuming that the melting enthalpy and melting entropy of per mole repeating unit of the polymer did not vary with the temperature, the thermodynamic properties of the 2-component polymer melts could be described by Nishi-Wang equations (Eq. [3]) (28, 29).
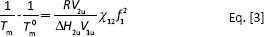
Ignoring the effects of entropy, the interaction energy density B for the characteristics of polymers is descried by the Equation [4].

In Equations [3] and [4], V1uand V2u are the molar volume of 2 component repeating units (cm3 mo1-1); ∆H2u is the melting enthalpy of per molar repeating units (kJ mol-1); χ12 is the interaction parameter of polymer-polymer, the melting point can reduce only if the value is negative; φ1is the volume fraction; Tm is the equilibrium melting point of polymer in blend (oC) and Tm0 is the equilibrium of neat polymer (oC).
Thus, there is a linear relationship between (1/Tm-1/Tm0)/φ1 and φ1/Tm (Eq. [5]). The drawing was made with (1/Tm-1/Tm0)/φ1 as ordinate and φ1/Tm as abscissas, and the curve was linearly fitted as shown in Figure 9.
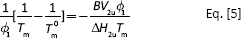
If B or χ12 is independent of the composition, it can get a straight line, and the interaction energy density B and interaction parameter χ12 in the blending system could be calculated by the slope of linear. However, the fitting curve of the PLLA/PLL blend system was not a strictly straight line (Fig. 9), which indicated that either B or χ12 was relevant with the composition. The fitting slope of the curve was −1.03, which meant either B or χ12 was negative and indicated the PLLA/PLL blends were a compatible system of thermodynamic stability in a molten state. That is to say, there exist close interactions between the PLLA and PLL molecules in the blend system. It is shown that the generated copolymer could lead to the melting point of neat component decline if an ester exchange reaction emerged from the crystalline polymer blends (30). Since PLLA and PLL molecules contain a large number of ester groups in the blend, ester exchange reactions between PLLA and PLL molecules occur during the melting, which could contribute to their compatibility and cause the lowering in the PLLA melting point.
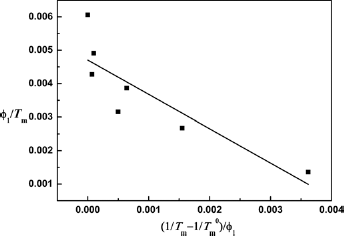
Fitting curve of (1/Tm-1/Tm0)/φ1 - φ1/Tm of the PLLA/PLL blend system.
The melting point of the polymer and the size of lamellar crystal were related to the intergrality of crystallization (19). The addition of PLL affected the crystallization of PLLA, which led to the imperfection of the PLLA wafer. This was the cause of the melting point decrease in the morphology structure.
Crystallization morphologies of PLLA/PLL blends
The crystalline grain size, crystallinity, crystallization velocity and crystallization temperature of the polymer had significant influence on its properties. The POM photos of PLLA and PLLA/PLL blends are shown in Figure 10. PLLA spherulites in PLLA or PLLA/PLL blends met each other and were truncated to irregular polyhedra when they were growing from the cooling melts. In PLLA/PLL blends, amorphous PLL dispersed in the PLLA spherulites (Fig. 10b, c, d and e) with the approximate spherical nanoparticle form. With the increase of PLL content in the blends, the amorphous PLL aggregated to larger phase dispersing among the PLLA spherulites, causing the dark fields inside the PLLA spherulites to become larger, the crystallization area to shrink, and PLLA spherulites to retain only the outlines of the circular spherulites. PLLA/PLL blends presented uniform morphology structures macroscopically but actually the phase separation occurred microcosmically. That is, PLLA/PLL blends were partially compatible from morphological structures (31).

Polarizing optical microscopes of crystallization morphology of PLLA and PLLA/PLL blends (cooling rate of 3 oC 5min-1). (
The proportion of crystalline/amorphous component in the blend had a significant effect on the morphology of the system. From Figure 10, the spherulite morphology of PLLA in PLLA/PLL blends did not alter after the addition of PLL, but the size and arrangement of spherulite changed. The crystallization morphology of PLLA was dominated by the spherulites, which were formed by the crystallization of flake folding chains. The spherulite morphology was relatively complete and the PLLA spherulites stacked closely together, crowding each other. The apparent Maltese cross appeared in the vibration direction of the polaroid and the remaining light distribution generated by the refraction was uniform (Fig. 10a). When the PLL content in PLLA/PLL blends was less than 30 wt%, the morphology of the PLLA spherulite appeared as a circular and Maltese cross, the spherulites almost filled in the whole blends and the crystalline regions were continuous. With the increase of PLL content in the blends (more than 50 wt%), the crystalline phase was no longer continuous gradually but the amorphous regions became continuous (Fig. 10d, e). If the 2 kinds of crystalline/amorphous blending polymers are incompatible, the crystalline polymer will crystallize independently according to the crystallization behavior of individual, owing to the strong repulsion between amorphous polymer molecules and crystalline polymer molecules. However, the amorphous PLL entered into the lamellaes of the PLLA spherulites, and the existence of PLL affected the PLLA crystallization morphology in PLLA/PLL blends, which indicated that the PLLA and the PLL were partially compatible. The crystal structures of PLLA and PLLA90/PLL10 films were investigated by XRD.
Figure 11 is the XRD curves of PLLA90/PLL10 and PLLA films. From the figure, PLLA showed sharp peaks at 2θ 16.9o, 19.3o and 22.4o, which were the typical diffraction characteristic peaks of crystalline polymer. The XRD peaks of PLLA90/PLL10 were at 2θ 14.6o and 22.3o, and there was only a weak broad peak near 19.0o. The blending with PLL made the PLLA diffraction peaks wide and dispersed. These results indicated that the addition of PLL reduced the regularity of the arrangements of PLLA chains, and PLL changed the lattice parameter and crystalline structure of PLLA and decreased the crystalline behavior of PLLA.
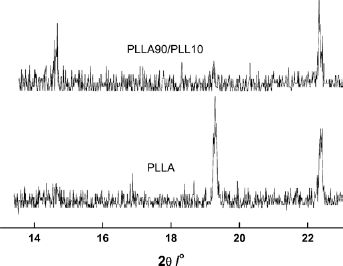
XRD curves of PLLA90/PLL10 and PLLA films.
The thermal and crystallization properties of the PLLA/PLL blend indicated that the PLLA had a certain degree of miscibility with the PLL. There did not appear significant phase separation when the content of PLL in the blend was lower than 50 wt%. It was believed that phase separation was liable to occur and the interface was clear with poor compatibility in the blend of the PLLA and amorphous PDLLA (17). The reasons why the PLLA were partially miscible with the amorphous PLL by contrast with the PDLLA is likely that the part amino groups and hydroxyl in the molecular chains of PLL form a hydrogen bond with the carbonyl in PLLA, which enhances the intermolecular forces. The phase separation between the PLLA and PDLLA, on the other hand, occurred due to the strong repulsion force between molecules. Moreover, the relative molecular mass of PLL was lower than that of PDLLA which made PLL disperse easily into the PLLA molecular chain gap. Nevertheless, the PLLA and the PLL were not completely miscible because partial phase separations appeared in the blends. The main reason was that the PLL was hydrophilic but PLLA was lipophilic. The POM results of the PLLA/PLL blends agreed with the DSC results.
The sizes of the PLLA spherulites increased with the increase in PLL content but decreased when the content of PLL in the blend was more than 50 wt%. The PLL played the role as diluent in the blend and provided an active environment for the PLLA segments when the content of PLL in the blend was below 50 wt%, so the size of the PLLA spherulite enlarged to alleviate the close packing of the spherulites. When the PLLA was diluted to a certain extent, the PLLA spherulites even stopped growing due to the lack of molecular chains, so the spherulite sizes decreases gradually. The imperfect structures of the PLLA spherulites and the decrease of crystallinity were beneficial for improving the impact ductility.
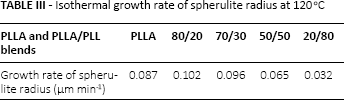
From Table III, the growth velocity of PLLA spherulites changed from fast to slow with the increase of the PLL content. When the content of PLL in the PLLA/PLL blend was lower than 30 wt%, the crystals grew rapidly because the PLL molecules entered into the PLLA molecular chains, reducing the intermolecular interaction and making the PLLA molecular chains compliant. However, when the PLL content was higher than 30 wt%, a large amount of PLL hindered the rearrangement of the molecular chains of PLLA, making it difficult to grow into wafers and form into spherulites.
Isothermal growth rate of spherulite radius at 120 oC
Analysis of the hydrophilic properties of the PLLA/PLL blend
PLL with hydrophilic amino groups is a kind of hydrophilic/lipophilic polymer while PLLA is a lipophilic polymer. The hydrophilic properties of PLLA/PLL blends can affect the degradation periods in an aqueous environment. The variation of the hydrophilic properties of PLLA/PLL blends was evaluated by measuring the contact angle of water with the surfaces of PLLA/PLL hot-pressing sheets. Figure 12 is the curve of water contact angles of PLLA/PLL blends and PLLA.
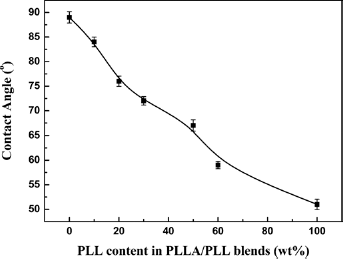
Water contact angle curve of PLLA/PLL blends and PLLA.
From Figure 12 it can be seen that the contact angles decreased with the increase of PLL content in the blends. That is, the hydrophilic property of PLLA was improved by the blending with the PLL. The hydrophilic/lipophilic properties could be adjusted to control the degradation by modifying the blend composition.
Tensile properties of the PLLA/PLL blend
The tensile results of PLLA and PLLA/PLL blend membranes are shown in Figure 13. Both tensile strength and elongation at break of the blends were higher than that of the pure PLLA when the content of PLL was less than 10 wt% in the blend. Notably, the tensile strength of PLLA90/PLL10 increased by 12.7 wt% and the elongation at break by 24.5 wt% compared with the pure PLLA. The mechanical properties of the blends were mainly provided by the PLLA with long molecular chains. The reason for the increase of tensile strength was that there existed strong interactions and hydrogen bonds between the PLLA and PLL, causing the phase domain to be smaller and the phase interface to be ambiguous, so the phase bonding between PLLA and PLL increased. The blends became more compact as the PLL of low relative molecular mass dispersed into the PLLA molecular chain gaps. Moreover, the decrease of crystallinity led to the increase of toughness. However, when the content of PLL was higher than 20 wt%, the tensile properties of the blends showed a downward trend with the content increase of the low average molecular weight PLL.
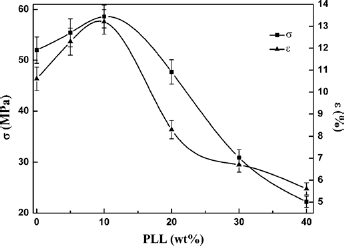
Tensile properties of PLLA and PLLA/PAL blends; σ is the tensile strength and ε is the elongation at break.
Figure 14 is the SEM micrographs of the tensile fracture surfaces of PLLA90/PLL10, PLLA and PLLA80/PLL20. The microstructure of PLLA90/PLL10 fracture surface was uniform and dense, and the fracture surface had no obvious phase interface. The fracture surface presented typical ductility characteristics. The matrix showed large yield deformation and there were drafting structures throughout the fracture surface, dissipating lots of energy and improving the toughness. The PLLA was a typical brittle material. The toughness of PLLA material was improved by blending with the 10 wt% PLL. The fracture surface of PLLA slice presented as a peeled lamellar structure with small amounts of crazing, which indicated that the fracture of PLLA approached the brittle fracture. Ductile tearing did not appear in the fracture surface of PLLA80/PLL20, and the fracture surface was a brittle structure without plastic deformation.

SEM micrographs of the tensile fracture surfaces of PLLA90/PLL10, PLLA and PLLA80/PLL20. (
Conclusions
In this study, synthesized PLL was an amorphous and hydrophilic/ lipophilic polymer, and it was used as a modification agent to PLLA by blending. The melting point of the PLLA/PLL blending system decreased with the increase of PLL content, while the crystal imperfection of PLLA increased. The crystallinity of PLLA in the blends varied with the component content, which showed that the amorphous PLL in the blend affected the crystallization behavior of PLLA. The thermal properties and crystalline properties of the PLLA/PLL blends indicated that PLLA and PLL had partial miscibility. The phase separation did not occur in an obvious fashion and the PLLA/PLL blends demonstrated increasing tensile properties and toughness when the content of PLL was less than 10 wt%. The hydrophilic property of the blend was improved with the increase of PLL content in PLLA/PLL blends because of the amino groups in PLL.
Footnotes
Financial support: Grants were received from the Northwestern Polytechnical University Fundamental Research Fund (JCY20130148) and the Seed Foundation of Innovation and Creation for Graduate Students in Northwestern Polytechnical University (Z2015030).
Conflict of interest: None of the authors has financial interest related to this study to disclose.