
Abstract
“Ne è passata di acqua sotto i ponti.” It has been a long time since the diagnosis of Hodgkin lymphoma (HL) was exclusively based on the detection of typical Reed-Sternberg cells and the recognition of the characteristic morpho-histological background, as well as on the pathologist’s skill. The discovery of immunologic, molecular genetic and virologic biomarkers has provided an objective contribution to the diagnosis and a scientific basis for a modern classification of HL. Recent updates have clarified the nature of the so-called nodular lymphocyte predominant HL and its link to the T-cell/histiocyte-rich large B-cell lymphomas as well as its relationship with the lymphocyte-rich subset of classical HL (CHL). Molecular virology studies assessed a role for the Epstein-Barr virus in the pathogenesis of a fraction of CHL of the general population, and virtually in all cases of CHL occurring in people infected by HIV. Finally, immunologic and genetic findings corroborated the existence of grey zone lymphomas at the edges of CHL. Overall, these advances provided additional and useful information to address the treatment of patients affected by HL.
Keywords
Histologic classification of Hodgkin lymphoma (HL) evolved through different systems, starting from the histologic classifications by Jackson and Parker in 1944 (1) and Lukes and Butler in 1966 (2), to the 2008 and 2017 World Health Organization (WHO) classification (3, 4). HL has been classified into classical HL (CHL), which accounts for 95% of all HL cases, and the less common nodular lymphocyte predominant HL (NLPHL). CHL is a neoplastic entity with typical clinical and epidemiological characteristics, and heterogeneous pathological, genetic, immunologic and virologic features. HL classification has not changed over the last 2 decades; however, updates concerning HL subtypes have incorporated information with significant clinical and biological implications (5). Updates concerning HL particularly regarded NLPHL and lymphocyte-rich CHL (LRCHL). Moreover, an unclassifiable B-cell lymphoma category was acknowledged for cases showing features intermediate between diffuse large B-cell lymphoma of the primary mediastinal subtype and CHL (5). We wonder if updates concerning HL really impact classification, diagnosis, and management of affected patients. In this article we try to give an answer.
Recent immunophenotypic and genetic data have convincingly indicated that the so-called NLPHL may be more precisely classified as a B-cell lymphoma with a follicular germinal center cell origin (6-9), and therefore pulled out from HL classification. In NLPHL, the lymphocyte predominant tumor cells display B-cell associated antigens, in most instances do not co-express CD30 and CD15, and are not permissive for Epstein-Barr virus (EBV) infection. However, there are specific works from the United States National Cancer Institute group describing the expression of all of these markers separately in NLPHL (10, 11).
LRCHL has features that are intermediate between NLPHL and other subtypes of CHL (5), in which tumor cells co-express CD30 and CD15; in part or completely lack B-cell differentiation; and may be infected by EBV. Therefore, cases of LRCHL morphologically resembling NLPHL in terms of nodular growth and lymphocyte richness could be correctly recognized as CHL on phenotypic grounds (12) (Fig. 1; Tab. I).
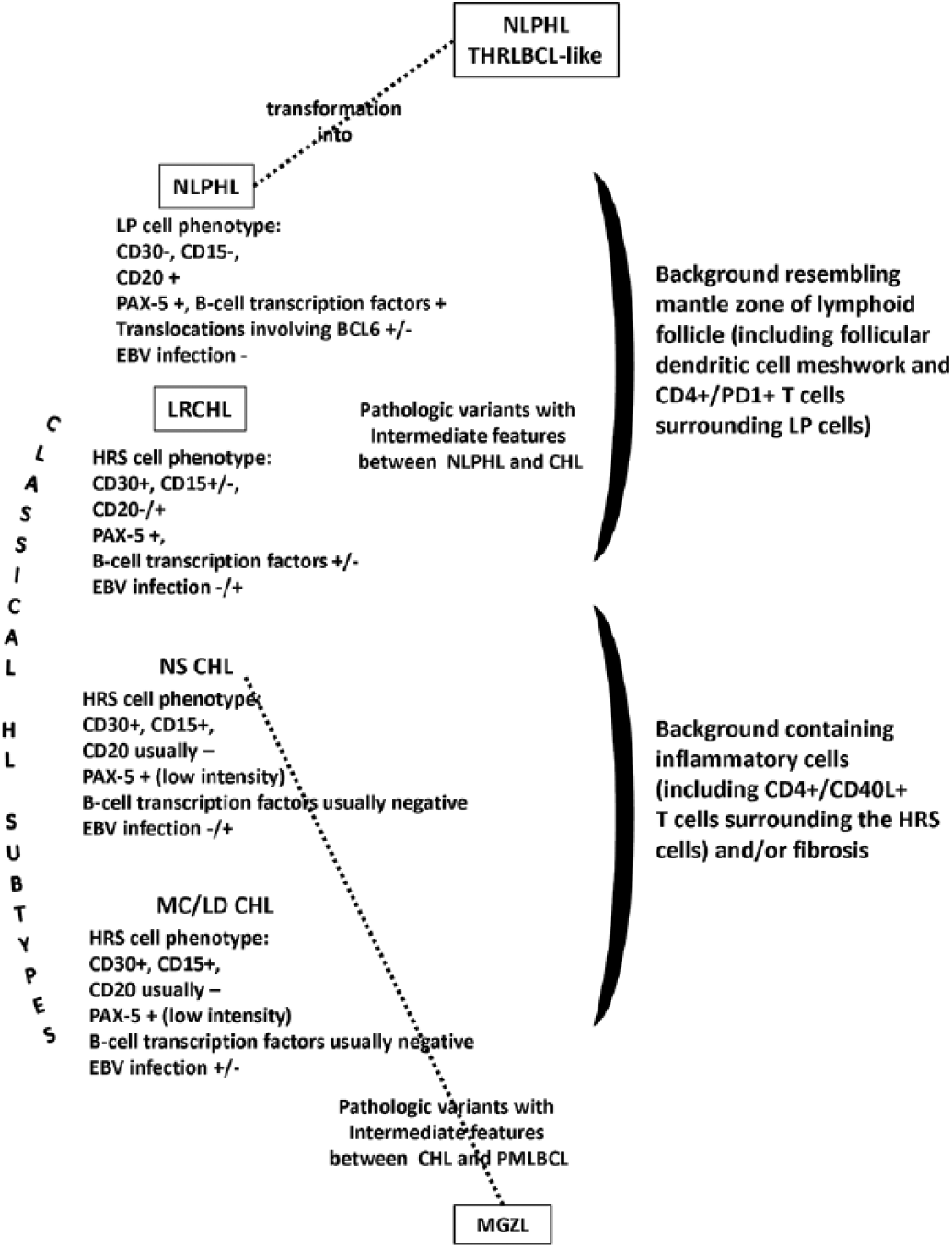
Hodgkin lymphoma classification after the revision of the WHO classification of lymphoid neoplasms (3-5). NLPHL has been separated from CHL subtypes, based on the expression of a B-cell phenotype and characteristic clinical behavior. The 2016 revision of the WHO classification acknowledged that NLPHL may evolve to a diffuse T-cell-rich proliferation that has been regarded as a THRLBCL-like transformation. LRCHL displays features intermediate between those of CHL and NLPHL. An unclassifiable B-cell lymphoma category, the so-called mediastinal grey zone lymphoma, was designated for cases with features between DLBCL of the primary mediastinal type (PMLBCL) and CHL, especially of the NS subytpe. While in NLPHL tumor cells, the LP cells are not permissive for EBV infection. EBV infection of HRS cells is a marker detectable in immunodeficiency-associated CHL (not shown) and, though less frequently, in CHL subtypes occurring in the general population. CHL = classical Hodgkin lymphoma; EBV = Epstein Barr virus; HRS = Hodgkin Reed-Sternberg; LD = lymphocyte depleted; LP = lymphocyte predominant; LRCHL = lymphocyte-rich CHL; MC = mixed cellularity; MGZL = mediastinal grey zone lymphoma; NLPHL = nodular lymphocyte predominant Hodgkin lymphoma; NS = nodular sclerosis; PMLBCL = primary mediastinal large B-cell lymphoma; THRLBCL = T-cell histiocyte rich large B-cell lymphoma; WHO = World Health Organization. Updated and/or confirmed entities are within squares.

CHL = classical Hodgkin lymphoma; DLBCL = diffuse large B-cell lymphoma; EBV = epstein-Barr virus; LRCHL = lymphocyte-rich CHL; NLPHL = nodular lymphocyte predominant Hodgkin lymphoma; NOS = Not otherwise specified; THRLBCL = T-cell histiocyte rich large B-cell lymphoma; WHO = World Health Organization.
Importantly, NLPHL may evolve to a diffuse T-cell-rich proliferation that has been regarded as a T-cell histiocyte-rich large B-cell lymphoma (THRLBCL)-like transformation (5). Such evolution is histologically and phenotypically indistinguishable from de novo THRLBCL. NLPHL that progress to a diffuse T-cell-rich pattern as NLPHL THRLBCL-like requires an aggressive treatment, because of the clinical association of this pattern (13). The rationale for adopting THRLBCL-like terminology in the updated WHO classification (5) relies on the recognition of overlapping NLPHL/THRLBCL entities (14), in addition to the recognition of variant patterns of NLPHL by Fan and colleagues (15) from the Stanford group, and then on the validation of the prognostic impact of variant patterns in NLPHL by the German Hodgkin Lymphoma Study Group data later (13).
The updated classification also acknowledged borderline categories for cases that do not clearly fit into 1 entity (5). An unclassifiable B-cell lymphoma category was acknowledged for cases showing features intermediate between primary mediastinal large B-cell lymphoma (PMLBCL) and CHL (16-20) (Fig. 1; Tab. I). In these cases - which were previously called mediastinal grey zone lymphoma (MGZL) (18-20) - Hodgkin Reed-Sternberg (HRS)-like tumor cells are admixed with large tumor cells with clear cytoplasm. The background usually contains an inflammatory infiltrate resembling that found in CHL. Sclerosis is variable and necrosis is frequent. In most cases, the diagnosis of B-cell lymphomas may be indicated by the absence of CD15 expression and the usual positivity for B-cell antigens and B-cell transcription factors. Clinical features, including age (young people), gender (males more than females), and site of presentation (mediastinal with limited disease), may also favor the diagnosis of MGZL (16, 17, 20). MGZLs are often found to be refractory to different chemotherapy regimens used in either DLBCL or CHL, such as CHOP-R, ABVD, and BEACOPP (17).
Unchanged issues in the revision of the WHO lymphoma classification (5) include immunodeficiency-associated CHLs; that is, HIV-associated CHL, Hodgkin-type post-transplant lymphoproliferative disorder and iatrogenic-methotrexate CHL, which were described within distinct chapters by the WHO Blue Book (4). Notably, immunodeficiency-associated CHLs have distinct epidemiological, biological, and clinical characteristics (21, 22). For example, EBV infection of HRS cells is a marker invariably or frequently detected in HIV-associated CHL and other immunodeficiency-associated CHL (21, 22).
Recognition of EBV+ DLBCL, Not otherwise specified, and EBV-mucocutaneous ulcer (designated as provisional entity) complicates the diagnosis in some instances, and awareness of these CD30+/EBV+ entities is necessary before calling a pathologic lesion CHL (e.g., in an enteric ulcer of a posttransplant patient). Clearly, the coexpression of CD30 and CD15 by typical HRS cells within an appropriate background rich in inflammatory cells, sclerosing collagen, and CD4+/PD1+ T-cells, provides a sharp separation between CHL and B-cell lymphomas or other pathologic conditions. Moreover, a distinction also may be based on EBV status, which is heterogeneous among the CHL subtypes and at variance with EBV+ DLBCL. In HIV-uninfected people, EBV may occur, but infrequently, in nodular sclerosis CHL and LRCHL; however, it is found in 20%-40% or more of mixed cellularity CHL and lymphocyte depleted CHL (21).
Taken together, recent updates concerning HL have clinical and pathological implications regarding not only how to diagnose and classify these lymphomas, but also how to treat those affected patients. For example, NLPHL, which is phenotypically distinguishable from LRCHL and the other subtypes of CHL, may evolve into a proliferation resembling THRLBCL (5). Such THRLBCL-like transformation is associated with an aggressive clinical course (13), is a distinct feature of NLPHL, and has not been acknowledged for CHL subtypes. Moreover, patients affected by MGZL usually exhibit worse outcomes than patients with PMLBCL (17, 20) despite pathologic features being intermediate between those specifically found in DLBCL and CHL. Regarding treatment, patients affected by CHL are successfully treated with ABVD (23), while patients affected by NLPHL with increased large cells, THRLBCL-like and THRLBCL are all effectively treated with CHOP-R. Notably, patients affected by MGZL are currently treated with EPOCH-R. Very recently, 3 cases of refractory MGZL were successfully treated with programmed cell death protein-1 blockade (24). In conclusion, awareness of these treatment options is necessary during the diagnostic workflow to know which distinctions really are relevant for therapy.
Footnotes
Disclosures
Financial support: Supported, in part, by an Institutional Grant from the Centro di Riferimento Oncologico for a project on “Molecular Pathology.”
Conflicts of Interest: A.C. is Chairman of the Italian TNM Staging Committee - UICC, and a member of the WHO IARC Monograph Working Group on Biological Agents, Lyon, 2009. No other conflict of interest.