
Abstract
Epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein and a member of the tyrosine kinase superfamily receptor. Gliomas are tumors originating from glial cells, which show a range of aggressiveness depending on grade and stage. Many EGFR gene alterations have been identified in gliomas, especially glioblastomas, including amplifications, deletions and single nucleotide polymorphisms (SNPs). Glioblastomas are discussed as a separate entity due to their high correlation with EGFR mutants and the reported association of the latter with survival and response to treatment in this glioma subgroup. This review is a comprehensive report of EGFR gene alterations and their relations with several clinical factors in glioblastomas and other gliomas. It covers all EGFR gene alterations including point mutations, SNPs, methylations, copy number variations and amplifications, assessed with regard to different clinical variables, including response to therapy and survival. This review also discusses the current prognostic status of EGFR in glioblastomas and other gliomas, and highlights gaps in previous studies. This serves as an update for the medical community about the role of EGFR gene alterations in gliomas and specifically glioblastomas, as a means for targeted treatment and prognosis.
Introduction
Epidermal growth factor receptor (EGFR) is a transmembrane tyrosine kinase located on chromosome band 7p12. It is a member of the ErbB family of receptors which consists of 4 related tyrosine kinases: EGFR (ErbB-1), HER2/neu (ErbB-2), Her 3 (ErbB-3) and Her 4 (ErbB-4) (1). EGFR can be activated by the epidermal growth factor (EGF), transforming growth factor α (TGFα) or several other ligands. Upon activation, EGFR receptor dimerization induces autophosphorylation of tyrosine residues on the receptor C-terminal domains, eliciting downstream protein activation (2). EGFR signaling leads to the activation of the MAPK pathway intracellularly, signaling of PI3K and src kinase and activation of STAT transcription factor (3). Downstream pathways lead to DNA synthesis and cellular proliferation (4). EGFR has been shown to be implicated in tumorigenesis as an oncogene, as well as in inflammation such as psoriasis, eczema and atherosclerosis (5, 6). EGFR alterations and mutations have been identified in cancer tissue including lung (7), head and neck (8), breast (9-11), gastrointestinal tract (12, 13) and brain, and a lot of research has been dedicated to correlating EGFR alterations with clinical outcomes.
EGFR is a potent oncogene, and it is not a surprise that neoplasms have developed a way to augment this gene’s activity. EGFR has been found to be modified in several ways in different grades of astrocytoma especially glioblastoma (GBM) by overexpression, amplification, deletion mutants and others. EGFR alterations promote proliferation, survival, angiogenesis and invasion. Hence, alterations may impact gliomagenesis, but their prognostic, diagnostic and therapeutic clinical relevance remain controversial. In GBMs, EGFR amplification has been shown to promote invasion, proliferation and resistance to radiotherapy and chemotherapy (14-17). Several trials have failed to find conclusive evidence of a relation between the different EGFR gene and protein changes and survival, and trials on targeted anti-EGFR therapy have yielded variable results (18-20). Some researchers attribute the inconsistent results of clinical trials to the fact that targeted drugs are not being specifically designed to inhibit the aberrations most common to gliomas (21). Nevertheless, EGFR gene and protein alterations are still being assessed for their prognostic significance especially in GBMs, among which 40% present with amplified EGFR, 60% with overexpression and 24%-67% with a mutated gene (22, 23). This review will focus on the different EGFR gene and protein alterations reported in gliomas, mainly GBMs, and their correlation with several clinical factors including survival and response to treatment.
Spectrum of identified EGFR alterations in all gliomas
Many EGFR modifications in gliomas have been reported in the literature, some of which are specific to GBM (Fig. 1). EGFR amplification was seen in 0-4%, 0-33% and 34%-64% of grade II, III and IV astrocytomas, respectively (24-35). Polysomy of the entire chromosome 7 is the most common EGFR copy number variation in all gliomas (21). Forty-four percent of patients with EGFR amplification had EGFR point mutations, mostly seen in the extracellular domain – e.g., A289 or R108 (21). In one of the studies, EGFR amplification was only seen in GBM and was associated with EGFRvIII, a deletion mutant, and high levels of EGFRv2, v3 and v4, different products of gene splicing (36). Other studies reported EGFR amplification in GBMs, anaplastic oligodendrogliomas (AOs) and anaplastic oligoastrocytomas (AOAs), while EGFRvIII was only reported in GBMs and AOs (24-35).
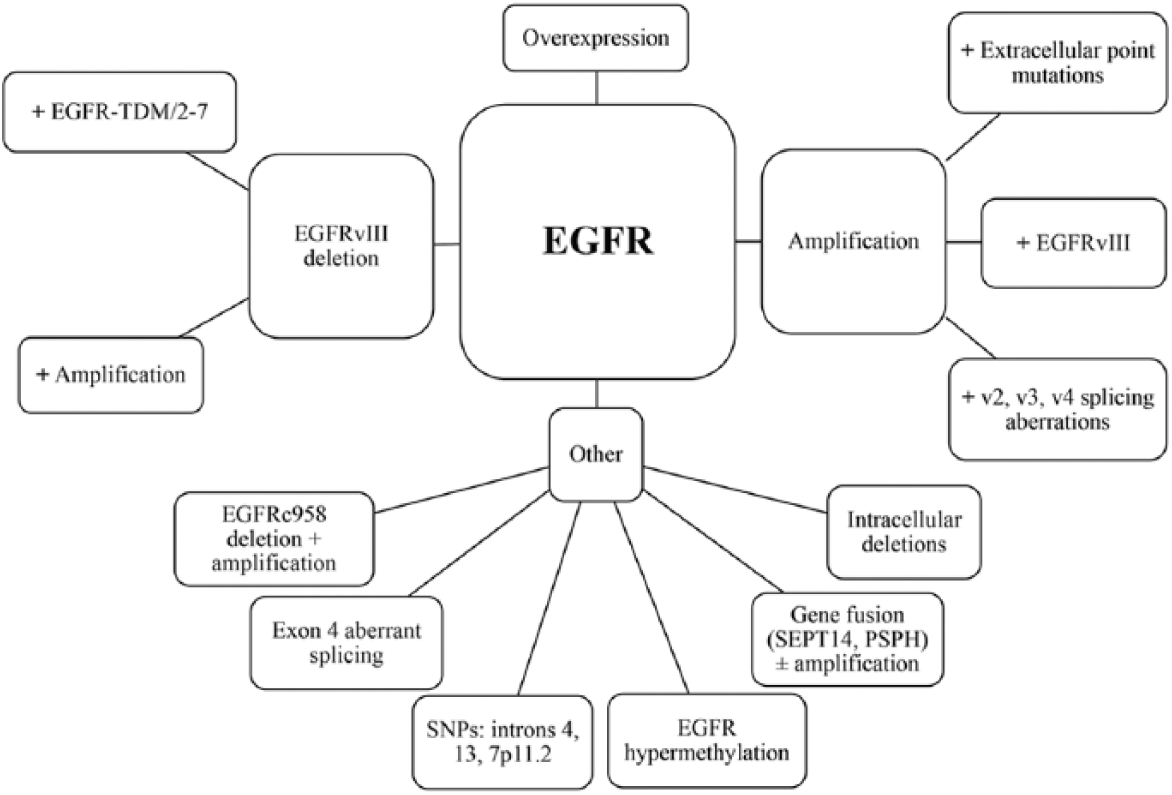
Epidermal growth factor receptor (EGFR) modifications in gliomas. SNPs = single nucleotide polymorphisms.
EGFR overexpression was seen in 6%-28%, 27%-70% and 22%-89% of grade II, III and IV astrocytomas, respectively, and represents an increase in gene transcription independent of DNA alterations (21, 24, 27, 28, 33, 37-60). EGFRvIII, an intragenic deletion of exons 2 to 7 affecting the extracellular domain, was tightly linked to EGFR gene amplification with some exceptions (24, 34), and was only found in high-grade astrocytomas, namely GBMs (21). In that respect, EGFRvIII generally requires EGFR amplification but can rarely occur without gene amplification (61). A study by Nishikawa et al showed that 50% of EGFR amplified tumors have EGFRvIII constitutively active ligand-independent mutant receptor with impaired down-regulation (62). Another study reported the coexpression of EGFRvIII and EGFR-TDM/2-7 (in-frame tandem duplication of exons 2-7) (63).
Many other less-studied EGFR alterations have been reported in the literature. These alterations include carboxy-terminal EGFR intracellular domain deletions in exons 25-27, 27-28 and 25-28; EGFR gene fusion in GBM to intron 9 of SEPT14 or PSPH with intact tyrosine kinase domain, with or without gene amplification; EGFR hypermethylation in early clonal evolution and recurrence; 2 single nucleotide polymorphisms (SNPs) in introns 4 and 13 with higher risk for gliomas; 7p11.2 SNPs; EGFR variant with aberrant splicing of exon 4; EGFRc958 deletion in amino acids 521-603 with EGFR amplification; and other specific EGFR point mutations (21, 64-69). Of interest is the fact that 2 EGF-specific findings have been reported: a polymorphism in the 5’-untranslated region of EGF, with a higher risk of postoperative recurrence and shorter progression-free survival; and 7 EGF SNPs correlated with shorter survival: 2 in the promoter region and 5 in introns 5, 7, 10, 11 and 13 (68, 70).
EGFR pathways and mechanisms of resistance in GBMs
Binding to receptor tyrosine kinases activates PI3K, which in turn phosphorylates phosphatidylinositol 4,5-biphosphate (PIP2) to form phosphatidylinositol 3,4,5-triphosphate (PIP3). Activated PIP3 renders AKT active, promoting mTOR complex 1 and 2 activation. mTORC1 induces cellular growth by increasing anabolic processes of lipids, proteins and organelle production, while decreasing catabolic reactions. mTORC2 downstream signaling promotes cellular proliferation and survival. Regulation of this pathway is achieved by several proteins including PTEN, an inhibitor of PIP2 phosphorylation (71). While wild-type EGFR preferentially activates the mTORC1 pathway, EGFRvIII, the deletion mutant of EGFR, activates the mTORC2 pathway, possibly contributing to EGFR-targeted treatment resistance (72, 73).
EGFRvIII activates a downstream pathway similar to EGFR, promoting AKT phosphorylation and decreased levels of P27KIP1, a cell cycle regulator that inhibits G1 to S phase transition (74-76). In vitro studies using an EGFRvIII glioma cell line displayed increased expression of abnormal spindle-like microcephaly-associated (ASPM) protein and matrix metalloproteinase 13 (MMP-13) which promote neural stem cell regeneration and tumor invasiveness, respectively (77, 78). Up-regulation of vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8) via the nuclear factor kappa-light chain enhancer of activated B cells (NF-κB) pathway promotes angiogenesis leading to further GBM progression (79).
Several mechanisms of chemotherapy resistance have been reported in the literature. To start with, EGFRvIII mutation was been shown to regulate apoptotic proteins independent of EGFR expression via up-regulation of Bcl-XL in EGFRvIII-suppressed tumors. This mechanism could contribute to GBM recurrence in tumors with suppressed EGFRvIII expression (80, 81). Preclinical studies have already shown a significant proapoptotic effect using Bcl-XL antisense RNA, and its combination with EGFR-targeted tyrosine kinase inhibitors (TKIs) might prove superior to EGFR therapy alone (82, 83). Inda et al reported that IL-6 can act as a paracrine signal released by EGFRvIII expressing cells to activate EGFR-expressing cells promoting tumor cell proliferation in vitro (84). IL-6 was also shown to manipulate Bcl-XL and STAT3 in other tumor types, and Bcl-XL inhibition relieved IL-6-induced tumorigenic growth effects (84, 85). Furthermore, tumor heterogeneity and convergence of downstream signaling could also lead to treatment resistance. In that, several cellular subtypes are involved in GBM tumors, some of which are dependent on EGFR receptor function while others are not. Different receptor tyrosine kinases have been shown to be altered in GBM tumors, leading to downstream activation of the same proteins activated by EGFR, promoting survival regardless of EGFR inhibitors (84, 86-89). Inhibitors of the EGFR pathway, specifically PTEN, have also been shown to contribute to resistance seen in EGFR-targeted therapies. Besides permanently activating EGFR downstream signaling, PTEN-deficient cell lines were shown to manipulate autophagic pathways by preventing TKI-induced caspase activation via heat shock protein accumulation, namely αB-crystallin (89-92). Interestingly, increased proapoptotic effects have been shown in erlotinib and mTOR inhibitor therapy after inhibiting autophagy-specific events (90, 92). All in all, resistance mechanisms support combination therapy use in GBM patients to target evolving compensatory mechanisms, and avoiding single-drug regimens that address only 1 aspect of a whole protein interactome.
EGFR as a prognostic factor in GBMs
EGFR seems a promising therapeutic target in GBMs. Indeed, EGFR has been found to be amplified in 40%, overexpressed in 60% and deleted or mutated in 24%-67% of cases (22, 23, 70). EGFR amplification and overexpression showed a homogeneous distribution across the tumor tissue and were barely detectable in normal brain tissue, which gives an advantage to EGFR-targeted therapy (93).
In a study of 87 supratentorial GBM patients by Shinojima et al, multivariate analysis demonstrated that EGFR amplification was an independent, significant, unfavorable predictor for overall survival (OS) (p = 0.038, hazard ratio [HR] = 1.67) (24). In this study, EGFR gene status was a more significant prognosticator in patients younger than 60 years (p = 0.0003, HR = 3.15). While EGFRvIII alone did not predict OS, EGFRvIII together with EGFR amplification was an independent, significant, poor prognostic factor for OS (p = 0.0044, HR = 2.71). This finding indicates that EGFRvIII overexpression in the presence of EGFR gene amplification is a strong indicator of a poor prognosis and plays a pivotal role in the progression of gliomagenesis (24).
Further, Hobbs et al described a heterogeneous group of GBM patients. No correlation was found between EGFR amplification and survival. However, when patients were stratified into low to moderately amplified (EGFR to chromosome 7 ratio = 2-20) and highly amplified EGFR GBMs (EGFR to chromosome 7 ratio >20), longer survival was found in the highly amplified group compared with the nonamplified, and an even longer survival period compared with the low- to moderately amplified tumor group (94). In a study of 83 GBM patients, EGFR amplified tumors had a better prognosis when co-occurring with a homozygous CDKN2A deletion (HR = 0.12; p = 0.01). On the other hand, a worse outcome was seen in EGFR-amplified tumors with chromosome 7 polysomy (HR = 14.88; p = 0.01), in young patients (HR = 3.75; p<0.01) and in radiotherapy-treated patients (HR = 2.71; p = 0.03) (95). Patients with EGFR-overexpressed GBMs studied after surgery and postoperative radiotherapy had a lower survival compared with those lacking the overexpression (12.5 months vs. 17.5 months; p = 0.013) (96). Nevertheless, some studies still report opposite clinical outcomes. For instance, a better progression-free survival (PFS) (p = 0.006) but not OS (p = 0.12) was reported by Lv et al in purely EGFR amplified tumors with no other mutations (97).
EGFRvIII mutation was found in up to 45% of GBMs (24, 34, 45, 98, 99). EGFRvIII coexisted with EGFR amplification in 67% of GBM tumors with heterogeneous expression across the tumor tissue, as opposed to EGFR amplification and overexpression (66, 100). Heimberger et al reported that neither EGFR expression nor EGFRvIII predicted OS in GBM patients after resection. However, EGFRvIII was a negative prognostic marker for patients surviving at least 1 year (45). Moreover, a randomized phase II trial of recurrent GBMs showed an association between immunohistochemical detection of EGFRvIII and low PFS (101), while other studies have reported no association with survival (24, 34, 45, 98, 99), and still others have found EGFRvIII to be associated with a better prognosis (95, 102). Combining different genes and intracellular signaling, EGFRvIII mutation with gene amplification, coexpression of PTEN, and activation of AKT/MAPK and YKL-40 is associated with a negative prognosis (19, 24, 34, 45, 99). Interestingly, Pelloski et al, in studying the largest GBM cohort to date, reported that classic prognostic factors in GBM (age, scores on the Karnofsky Performance Scale and Radiation Therapy Oncology Group recursive partitioning analysis score) were less relevant in patients harboring the EGFRvIII mutation, which would add to the complexity of finding relevant factors predicting outcomes in such patients (99) (Tab. I). Many studies report contradictory evidence concerning EGFR amplification and vIII mutant as prognostic markers, and a conclusive consensus cannot be reached yet.
Overall survival (OS) for different EGFR gene and protein alterations when associated with other demographic, clinical or genetic variables

EGFR = epidermal growth factor receptor.
Most outcomes reported are based on single studies that require further validation of results.
EGFR status as a predictor of treatment response
EGFR gene and protein status has been studied as a predictor of response to different chemotherapy regimens (Tab. II). Starting with first-generation EGFR TKIs, a phase I clinical trial testing erlotinib alone in recurrent high-grade glioma patients showed a significant correlation of EGFR protein overexpression, EGFR gene amplification and phosphorylated Akt underexpression with response to erlotinib and/or longer time to progression (103). Indeed, low phosphorylated-Akt expression, assessed using immunohistochemistry, tended to be correlated with higher PFS in recurrent GBM patients treated with erlotinib (p = 0.07). In contrast, EGFRvIII expression was associated with poor PFS (p = 0.003), but no association was found with response to erlotinib (103). Responses to erlotinib and gefitinib were favorable in tumors expressing EGFRvIII and intact PTEN (19). Along the same line of thought, EGFR amplification was found to have no correlation with outcome, and there was no effective blockade of downstream signaling in 22 patients with recurrent GBMs treated with gefitinib for at least 5 days before tumor resection surgery after which gefitinib was resumed (104). Both studies show that intact downstream signaling is needed for favorable response to EGFR-targeted therapies: erlotinib and gefitinib. In addition to classical EGFR-targeted therapies, the relationship between second-generation EGFR TKIs and clinical outcome was examined (108). EGFR expression and EGFRvIII were not associated with outcomes, although there was a non–statistically significant trend between EGFRvIII and outcomes.
Association of EGFR gene and protein status with response to different chemotherapy regimens in glioblastoma
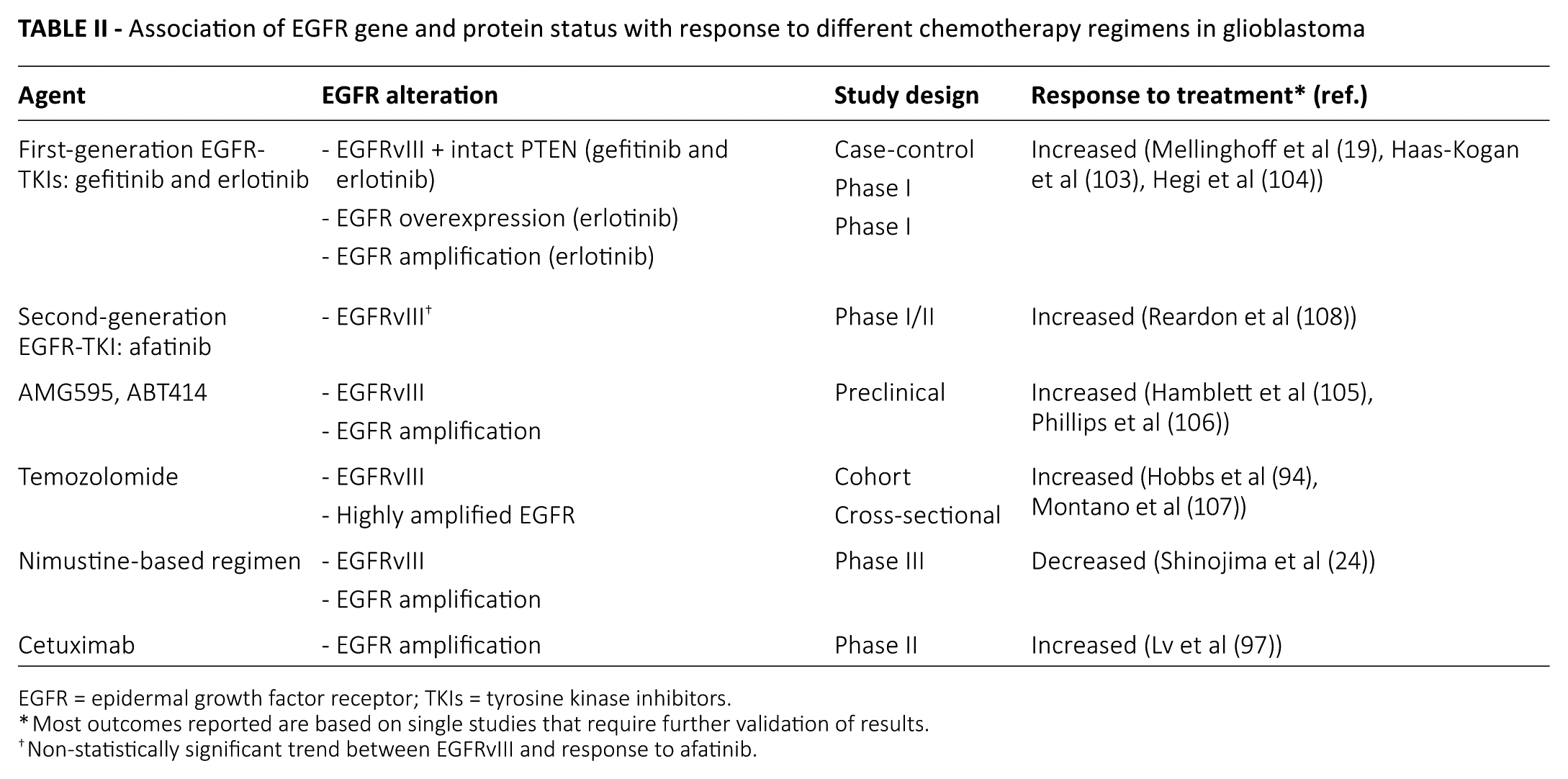
EGFR = epidermal growth factor receptor; TKIs = tyrosine kinase inhibitors.
Most outcomes reported are based on single studies that require further validation of results.
Non-statistically significant trend between EGFRvIII and response to afatinib.
EGFRvIII was associated with a prolonged survival in 73 GBM patients treated with temozolomide (TMZ) radiochemotherapy (107). When stratified into low- to moderately amplified (EGFR to chromosome 7 ratio = 2-20) and highly amplified EGFR GBMs (EGFR to chromosome 7 ratio >20), nonamplified and highly amplified tumors had a better response to TMZ compared with low to moderately-amplified tumors (94). Other treatments have shown some correlation with survival. Shinojima et al reported poor outcomes in GBMs with EGFRvIII overexpression and EGFR amplification in 87 patients on nimustine-based regimens (24).
Lv et al reported that patients with EGFR-amplified/ EGFRvIII-negative tumors had better outcomes with cetuximab in terms of PFS (p = 0.006) (97). In agreement with Lv et al, in a phase II trial, 90 patients with recurrent high-grade gliomas including 35 with GBMs were treated with cetuximab. PFS and OS were better in patients with EGFR amplified tumors, but the association did not reach statistical significance (109).
Many other studies have reported no correlation with EGFR gene and protein status. In a single-arm phase I/II study, erlotinib was given to 97 newly diagnosed GBM patients. Median OS was 15.3 months and did not correlate with EGFRvIII or EGFR amplification (110). Similarly, 97 patients were treated with gefitinib and radiotherapy without TMZ (111). Median OS was 11.5 months with no correlation with EGFR amplification or overexpression. The same results were reached in GBM patients treated with gefitinib alone when assessed for EGFR protein expression, EGFR amplification or EGFRvIII mutation (111-115). The addition of an mTOR inhibitor (everolimus) to gefitinib did not affect the relation between EGFR status and outcome (116). Likewise, no correlation with survival was found with EGFR or EGFRvIII in other studies of GBM patients treated with TMZ radiochemotherapy, erlotinib and sirolimus, or erlotinib and carboplatin (117-119).
It is important to note that response to specific small molecule kinase inhibitors such as gefitinib, erlotinib and second-generation TKIs do not directly parallel amplification and overexpression of EGFR, expression of the vIII mutant of EGFR, and increased phosphorylation of EGFR and its downstream signaling partners as shown by protein analysis, fluorescence in situ hybridization (FISH) and immunohistochemistry. Response to therapy may be predicted by an interplay between EGFR downstream signaling along with spatial filling into the tertiary kinase domain structure (120). Also, one cannot rule out the possible existence of several EGFR alterations that might modify the response to targeted therapies (61). Many clinical trials are currently being conducted relating EGFR alterations to chemotherapy treatment response (Tab. III).
Clinical trials relating treatment response to EGFR alterations
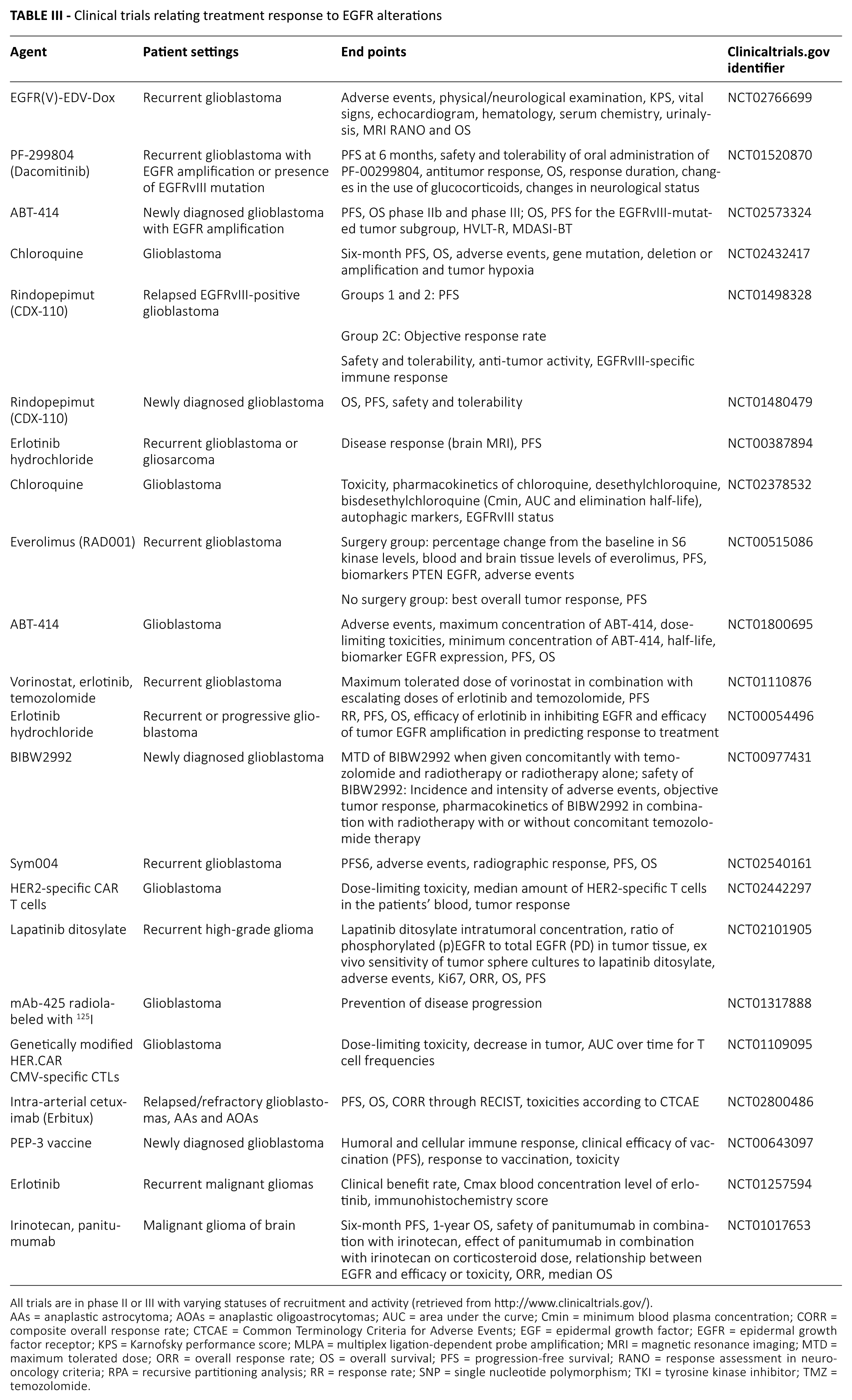
All trials are in phase II or III with varying statuses of recruitment and activity (retrieved from http://www.clinicaltrials.gov/).
AAs = anaplastic astrocytoma; AOAs = anaplastic oligoastrocytomas; AUC = area under the curve; Cmin = minimum blood plasma concentration; CORR = composite overall response rate; CTCAE = Common Terminology Criteria for Adverse Events; EGF = epidermal growth factor; EGFR = epidermal growth factor receptor; KPS = Karnofsky performance score; MLPA = multiplex ligation-dependent probe amplification; MRI = magnetic resonance imaging; MTD = maximum tolerated dose; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; RANO = response assessment in neuro-oncology criteria; RPA = recursive partitioning analysis; RR = response rate; SNP = single nucleotide polymorphism; TKI = tyrosine kinase inhibitor; TMZ = temozolomide.
Recent preclinical and clinical trials on anti-EGFRvIII and anti-EGFR antibody drug conjugates, such as AMG595 and ABT414, respectively, have shown some promising results for patients harboring these alterations (105, 106). Many trials have tested monoclonal antibodies to EGFR, resulting in diverse outcomes. A phase II trial on cetuximab, a mouse-human chimeric antibody, showed limited activity in the chosen population with high-grade gliomas (109). Nimotuzumab, a human antibody, showed some promising results in MGMT-negative, EGFR-amplified, non-completely resected gliomas and juvenile pontine gliomas as part of a combination therapy with radiation and vinorelbine, a vinca alkaloid (121). A phase II study on 125I-425 mAb, a radiolabeled murine antibody, reported that a combination with TMZ is highly promising as compared with control groups, and a phase III study is currently in progress (14). Another phase II study (http://clincialtrials.gov/; study no. NCT01017653) conducted on the human antibody panitumumab in combination with irinotecan, a topoisomerase I inhibitor, was prematurely terminated with no reported results.
Few studies have addressed vaccine therapy targeting EGFR alterations. In patients treated with rindopepimut (PEP-3-KLH), an EGFRvIII peptide vaccine, serum anti-EGFRvIII and EGFRvIII delayed-type hypersensitivity predicted OS (p<0.04) in a single-arm phase II trial of 21 GBM patients (122). However, recent phase III trial data show that survival outcomes did not improve in patients with EGFRvIII mutations (123). The ACT IV phase III trial of rindopepimut showed a higher median OS in the control arm (21.1 months) than the rindopepimut arm (20.4 months). Despite the negative results of this trial, another trial of rindopepimut (the ReACT trial) is ongoing for recurrent GBMs (123).
Another mechanism that can achieve EGFR inhibition is interference in genetic transcription or translation through antisense RNA, RNA interference (RNAi) and ribozymes (124). For instance, EGFRvIII-targeted antisense RNA could cause a decrease in tumor growth in an orthotopic xenograft model of human GBM (125). Using EGFR-targeted small interfering RNA (siRNA), OS increased to around 90% in an intracranial xenograft model of glioma, which confirmed the reduced proliferation seen in U251 glioma cell culture with EGFR-targeted siRNA knockdown (126). However, siRNAs are not as safe as other therapies. To enhance the safety of siRNA therapy, several modifications were implemented to address this matter – e.g., lowering RNA dosages and lengthening their sequences to target more upstream components of the siRNA pathway at lower doses, instead of higher doses more downstream in the pathway (124). Other available methods are to combine siRNA therapy with a vehicle (cyclodextrin-modified dendritic polyamine complexes [DexAMs]) or use codelivery with erlotinib (127). Another interference with translation is the use of anti-EGFRvIII hairpin ribozymes which cleave RNA substrates (128). MicroRNA-based therapies using the miR-7 inhibitor of the EGFR pathway have also been proven effective in cell culture studies (129). However, neither ribozymes nor microRNA methods have reached in vivo or clinical trials.
EGFR in other gliomas
EGFR amplification is rare in low-grade gliomas (130-132). Although it is rare, many studies have reported EGFR amplification varying in ranges of 0-4%, 0-33% and 34%-64% in grade II, III and IV astrocytomas, respectively. This amplification correlated to the histological malignancy grade and lower OS (24-35). The high prevalence in grade IV tumors reflects the significant role of EGFR amplification in the molecular pathogenesis of primary and secondary GBMs (133). Poor prognosis was seen in young individuals, while more amplifications and a better prognosis were seen in older patients (24-26, 30-32, 34, 134). This difference might explain an age-related variation in gliomagenesis. At the protein level, overexpression can range from 6% to 89% of reported cases of glioma, due to unknown mechanisms (24, 27, 28, 33, 37-60, 130, 132, 135-137). The EGFRvIII mutation was found in 8%-14% of anaplastic astrocytomas and 0% of oligoastrocytomas (34, 98). EGFR amplification and EGFRvIII mutation were independently associated with a lower survival in anaplastic astrocytoma patients, especially EGFRvIII in older patients (26, 34, 57, 98).
EGFR overexpression was seen in 6%-28%, 27%-70% and 22%-89% of grade II, III and IV astrocytomas, respectively (24, 27, 28, 33, 37-60). Overexpression was associated with tumor grade and with PFS and OS (24, 33, 38-40, 42, 44, 49-51, 53, 56, 57). On another note, a combination of EGFR overexpression and p53 mutation was associated with a shorter survival in a population consisting of glioma patients (133).
EGFR status and other associated factors
In addition to survival and treatment response, several other factors were associated with EGFR gene and protein status in glioma patients, with very limited information reported in the literature. In a population consisting of adult patients with anaplastic astrocytoma, AO or AOA, Yang et al reported a higher proportion of EGFR amplification, frontal lobe involvement, left cerebral hemisphere involvement and lower Ki-67 expression in patients with preoperative seizures in both univariate and multivariate analyses (138). Aghi et al reported that GBM-overexpressing EGFR had an increased T2/T1 ratio and a decreased T2 sharpness of the borders (139).
Conclusions
It is still early to disregard EGFR as a therapeutic and prognostic target in gliomas, especially GBMs, given the many limitations. Conflicting results seen on this topic are due to study limitations, including the retrospective nature of the studies, small study populations and inconsistent genetic detection methods (24, 107, 140). The conclusion that EGFR TKIs and other EGFR-targeted therapies are of no value is premature, due to the following characteristics of current TKIs: inadequate tissue penetration (141, 142), inadequate target inhibition (141), ineffective suppression of downstream signaling and redundant signaling pathways (61), alternative compensatory mediators of signaling (143, 144) and cellular heterogeneity (88, 89, 145). In addition to modifying EGFR-targeting drugs to overcome the above problems, drug testing must include the evaluation of downstream signaling upon treatment, assessment of drug delivery and the examination of dosing and combinatorial regimens (104, 146). Given the specificity of the EGFRvIII mutant and its heterogeneity in GBMs, the extent of EGFRvIII positivity can also be assessed regarding its prognostic significance and treatment response. Since classic prognostic factors have been shown to be irrelevant in the context of EGFRvIII mutant tumors, studies should address other markers and means to predict survival in these patients. In addition, more studies should address the potential of minor EGFR gene alterations, such as SNPs, as prognostic markers in glioma patients.
It is starting to become clear that EGFR is proving not to be an independent factor in gliomagenesis but a gene in a big hub of interacting genes leading to tumor formation. Although EGFR status as a clinical marker remains controversial, the aforementioned study limitations must be considered to come up with consistent results and better drug pharmacology. Finally, the need for larger-population studies is inevitable to examine the prognostic significance of EGFR gene and protein status for survival, treatment and other clinical factors affecting the patient’s outcome and quality of life.
Footnotes
Disclosures
Financial support: No grants or funding have been received for this study.
Conflict of interest: All authors declare they have no conflict of interest.