
Abstract
Background
Epidermal growth factor receptor (EGFR)–tyrosine kinase inhibitor (TKI) has demonstrated a promising therapeutic response in lung adenocarcinoma patients with EGFR gene mutations. However, the predictive factors for this therapy have not been established, except for the EGFR gene mutation status of carcinoma cells.
Methods
We first performed microarray analysis in EGFR-TKI–sensitive lung adenocarcinoma cell lines. The results indicated anterior gradient 2 (AGR2) as a potential surrogate marker of EGFR-TKI. Therefore, we then evaluated the correlation between the status of AGR2 immunoreactivity and clinicopathological factors including overall survival (OS), progression-free survival (PFS) and clinical response to EGFR-TKI, in 147 cases of surgically resected lung adenocarcinoma. The biological significance of AGR2 was further evaluated by transfecting small interfering RNA (siRNA) against AGR2 in these cells.
Results
The status of AGR2 immunoreactivity was significantly higher in lung adenocarcinoma cases with EGFR gene mutations than in those with the wild type (p<0.0001), but there were no significant differences in OS, PFS and response of EGFR-TKI between the AGR2 high and low carcinoma cases. Knockdown of AGR2 gene expression following siRNA transfection resulted in a significantly lower response to EGFR-TKI in EGFR-mutated PC-3.
Conclusions
AGR2 could serve as an adjunctive surrogate protein marker possibly reflecting EGFR gene mutations in lung adenocarcinoma patients. Results from in vitro analysis indicated that AGR2 could be a potential clinical biomarker of EGFR-TKI therapeutic sensitivity in lung adenocarcinoma cells.
Keywords
Introduction
Lung cancer is one of the most common malignancies in the world. Among different histological subtypes, adenocarcinoma accounts for approximately 30% of all lung cancer cases (1). Cytotoxic chemotherapy has been administered to advanced cases, but the response rate was 30.6% of all of those treated, and the median overall survival (OS) time of these patients was 12.6 months (2). Epidermal growth factor receptor (EGFR)–tyrosine kinase inhibitor (TKI) has recently demonstrated a remarkable therapeutic response in some non-small cell lung cancer (NSCLC) patients (3). In particular, the reported clinical response rate of EGFR-TKI in advanced lung adenocarcinoma patients with EGFR gene mutations was 73.7%, with a median OS time of 30.5 months with crossover standard cytotoxic chemotherapy (4). EGFR-TKI therapy has demonstrated great efficacy in patients with EGFR gene mutations in their carcinoma cells (5, 6). Thus, the most important predictive factor for the efficacy of EGFR-TKI is EGFR gene mutation status, which has been incorporated into the routine practice of diagnostic laboratories (7, 8). Among patients with EGFR gene mutations, the response rate is particularly higher in those with a deletional mutation in exon 19 or a L858R point mutation in exon 21, than in those with other types of mutations (9). These mutations have been detected in 90.9% of all mutation-positive NSCLC patients (9).
In addition, various factors predicting clinical resistance to EGFR-TKI have been reported in the literature. An insertion mutation (T790M) in exon 20 was first reported as a mechanism of acquired resistance (10). The presence of MET gene amplification in carcinoma cells was then reported to result in the development of clinical resistance through the promotion of ERBB3 (HER3) signaling (11). Hepatocyte growth factor (HGF), a ligand of MET oncoprotein, was then reported to contribute to therapeutic resistance to EGFR-TKI – in particular, to intrinsic resistance (12). However, the detailed mechanisms of therapeutic resistance to EGFR-TKI in NSCLC patients associated with EGFR gene mutations have not been clarified. In addition, if the efficacy of EGFR-TKI could be more precisely estimated before treatment, therapy more tailored to individual lung cancer patients could become clinically feasible. Therefore, it has become of pivotal importance to identify surrogate markers of EGFR-TKI, which could further refine the current screening of lung adenocarcinoma patients based on EGFR gene mutation analysis in carcinoma cells. We previously reported that the amounts of carcinoembryonic antigen-related cell adhesion molecule (CEACAM) family 3, 5 (also known as CEA), 6, 7 and 19 were significantly higher in EGFR gene mutated adenocarcinoma cases than in those with the wild-type EGFR gene (13). These CEACAMs were initially revealed by a microarray analysis in several EGFR-TKI–sensitive lung adenocarcinoma cell lines. The results of the in vitro EGFR-TKI sensitive/microarray analysis also revealed that anterior gradient 2 (AGR2, also known as AG2, HAG-2 or GOB-4) could be a candidate surrogate marker of EGFR-TKI sensitivity (Supplementary Figure 1 - Available online at www.biological-markers.com). AGR2 expression has been reported in several adenocarcinomas (14), including breast (15), prostate (16) and lung adenocarcinomas (17, 18). AGR2 was also reported to inhibit p53-induced activation in UV-irradiated H1299 cells (19). However, whether AGR2 is involved in the sensitivity of EGFR-TKI in lung adenocarcinoma patients remains largely unknown.
Therefore, in this study, we first studied the status of AGR2 in 147 surgically resected lung adenocarcinoma cases. The correlation between the status of AGR2 and clinicopathological factors including stage, OS, progression-free survival (PFS), Ki-67 labeling index (LI) in carcinoma cells (20), EGFR gene mutation and EGFR-TKI response were then evaluated. We also examined the effects of AGR2 on cell proliferation and the response to EGFR-TKI in lung adenocarcinoma cell lines, especially with relation to the EGFR gene mutation status.
Materials and Methods
Patients and Tissue Specimens
A total of 147 cases of lung adenocarcinoma were included from patients who underwent surgical resection from 2000 to 2006 in the Department of Surgery, Tohoku University Hospital, Sendai, Japan, and Miyagi Cancer Center, Natori, Japan. The clinicopathological features of these cases are summarized in Table I. A total of 52 cases were EGFR gene mutation-negative or wild type, and 49 cases showed EGFR gene mutations identified by the peptide nucleic acid–locked nucleic acid (PNA-LNA) polymerase chain reaction (PCR) clamp method (21). The clinical response to EGFR-TKI (gefitinib/erlotinib) treatment and PFS in the patients were evaluated based on Response Evaluation Criteria in Solid Tumors (RECIST, version 1.0) criteria in 34 patients with EGFR gene mutations who had been treated with EGFR-TKI (22). The clinicopathological characteristics of these 34 patients are listed in Table II. Fifteen EGFR mutation-positive patients did not receive EGFR-TKI treatment during their clinical course. The research protocols for this study were approved by the ethics committees at Tohoku University School of Medicine (2009-380) and Miyagi Cancer Center (H24. no. 34) for the respective groups of patients. Informed consent was obtained from each patient before surgery.
Correlation between clinicopathological factors and status of agr2 immunoreactivity in 147 lung adenocarcinoma cases
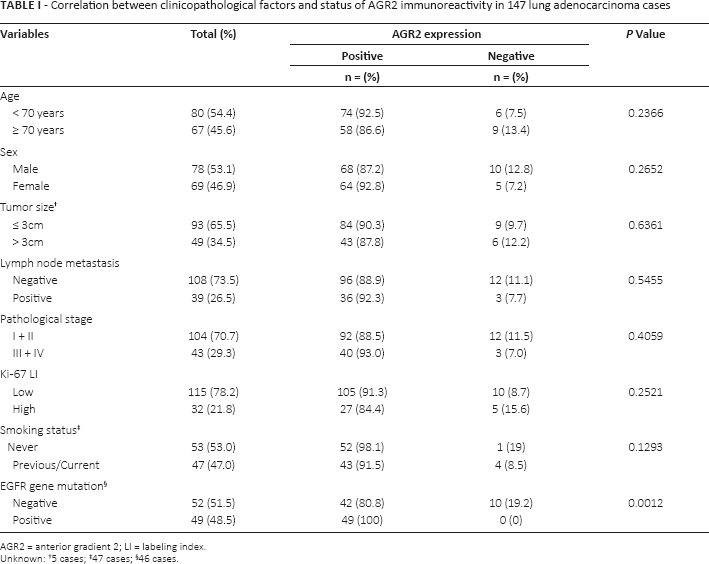
AGR2 = anterior gradient 2; LI = labeling index.
Unknown: †5 cases; ‡47 cases; §46 cases.
Clinicopathological factors of 49 patients harboring egfr gene mutation

EGFR-TKI = epidermal growth factor receptor–tyrosine kinase inhibitor; LI = labeling index.
Immunohistochemistry
The rabbit polyclonal antibody against AGR2 (NB110-17780) was purchased from Novus Biologicals (Littleton, CO, USA), the mouse monoclonal antibody against Ki-67 (MIB1) from DAKO (Carpineteria, CA, USA). Serial 4-μm-thick tissue sections were deparaffinized with xylene and ethanol. Antigen retrieval was performed by heating the slides in a microwave at 500 W for 20 minutes in citric acid buffer (pH 6.0) for AGR2, or by heating in an autoclave at 121°C for 5 minutes in citric acid buffer for Ki-67. Streptavidin biotin amplification method was employed for immunostaining using a Histofine Kit (Nichirei, Tokyo, Japan). After blocking nonspecific background immunostaining, the slides were incubated for 18 hours at 4°C with the primary antibodies. The dilutions of the primary antibodies were 1:200 for AGR2 and 1:100 for Ki-67. Endogenous peroxidase activity was blocked by immersing the slides in 0.3% hydrogen peroxidase for 30 minutes at room temperature. After incubation with the secondary antibodies, peroxidase-labeled streptavidin, antigen-antibody complex was then visualized with 3,3-diaminobenzidine (1 mmol/L, in 50 mol/L Tris–HCl buffer, pH 7.6, and 0.006% H2O2) and counterstained with hematoxylin. Normal human colonic mucosa was used as a positive control for AGR2 (23). As a negative control, an immunoabsorption test was conducted for the polyclonal antibody. The primary antibodies were replaced with normal rabbit nonimmune IgG for the monoclonal antibody. No specific immunoreactivity was detected in these preparations (data not shown).
Evaluation of Immunoreactivity
Immunoreactivity was examined independently by 2 of the authors (S.N. and Y.M.), who were blinded to the clinical data of the patients. AGR2 immunoreactivity was scored by immunoreactive score (IRS), by counting 1,000 carcinoma cells or more in each case. The percentage of immunopositive cells was scored as: 0 (0%), 1 (<10%), 2 (11%-50%), 3 (51%-80%) and 4 (>80%). The immunostaining intensity was also scored as: 0 (negative), 1 (weak), 2 (moderate) and 3 (strong) (18, 24). The percentage of positive cells and staining intensity was multiplied, resulting in a value between 0 and 12 (25). The cases were defined as positive when at least 10% of the tumor cells showed AGR2 immunoreactivity (the score of percentage ≥2) (17, 18). The Ki-67 LI was obtained by counting 1,000 carcinoma cells or more in each sample after identifying the hot spots under low-power magnification, and the percentage of immunoreactivity was subsequently determined as its LI (26). All of the cases examined were tentatively classified into 2 groups: the low Ki-67 group (LI <20%) and high Ki-67 group (LI ≥20%) (27, 28).
Cell Lines
In this study, we used A549, LCSC#1 and PC-3, which differs from PC-3 prostate cancer cell line (ATCC CRL-1435). The original tissues, sources and medium are summarized in Supplementary Table I, available online at www.biological-markers.com. The EGFR gene mutation, point mutation in exon 21 (L858R) and deletion mutation in exon 19 (L747-E749del A750P) were identified in LCSC#1 and PC-3, respectively, by PCR-Invader assay (BML, Tokyo, Japan). These cells were maintained in medium supplemented with 10% fetal bovine serum (FBS) (Nichirei).
Immunoblotting
Protein was extracted from A549, LCSC#1 and PC-3 using M-PER (Pierce Biotechnology, Rockford, IL, USA) with Halt Protease Inhibitor Cocktail (Pierce Biotechnology). Twenty micrograms of the protein was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 15% acrylamide gel). Following SDS-PAGE, proteins were transferred onto Hybond P polyvinylidene difluoride membrane (GE Healthcare, Chalfont Saint Giles, Buckinghamshire, UK). The primary antibodies used in this study were AGR2 (NB110-17780; Novus Biologicals) and β-actin (AC-15; Sigma-Aldrich, St. Louis, MO, USA). The dilutions of the primary antibodies were 1:500 for AGR2 and 1:1,000 for β-actin. Antibody–protein complexes on the blots were detected using ECL Plus Western blotting detection reagents (GE Healthcare). The protein bands were visualized with LAS-1000 image analyzer (Fuji Photo Film, Tokyo, Japan).
Quantitative Reverse Transcription Real-Time PCR
Total RNA was carefully extracted from A549, LCSC#1 and PC-3 using a TRIzol reagent (Invitrogen Life Technologies, CA, USA), and cDNA was synthesized using a QuantiTect Reverse Transcription kit (Qiagen, Hilden, Germany). RT-PCR was carried out using the Light-Cycler System (Roche Diagnostics, Manheim, Germany), and ribosomal protein L 13a (RPL13A) was also used as an internal standard. The PCR primer sequences used in this study were as follows: AGR2 (NM_006408), 5′-CCAGTATGTCCCCAGGATT-3′ and 5′-TCAGTCTTCAGCAACTTGAGAG-3′, and RPL13A (NM_012423), 5′-CCTGGAGAGAAGAGGAAAGAGA-3′ and 5′-TTGAGGACCTCTGTGTATTTGTCAA-3′. Negative controls, in which the reaction mixture lacked cDNA template, were included in this assay to exclude the possibility of exogenous, contaminant DNA. cDNA of known concentrations for AGR2 and the housekeeping gene RPL13A were used to generate standard curves for quantitative real-time PCR (qRT-PCR) to determine the quantity of the target cDNA transcript. The mRNA level in each case is represented as a ratio of RLP13A (%).
Small Interfering RNA Transfection and Gefitinib Sensitivity Test
Small interfering RNA (siRNA) oligonucleotide for AGR2 was purchased from Sigma-Aldrich. The sequence of siRNA against AGR2 was 5′-CUGAUUAGGUUAUGGUUUATT-3′. One scramble siRNA (Genolution Pharmaceuticals, Seoul, South Korea) was used as the negative control. Gefitinib was commercially obtained from Biaffin GmbH (Kassel, Germany). Ten nanomolar each of AGR2 and control siRNA were transfected in PC-3 using a G-fectin kit (Genolution Pharmaceuticals) according to the manufacturer's protocol. Cells were seeded in 96-well culture plates at a density of 3 × 104 cells/well after 48 hours of siRNA transfection. Following 24-hour incubation at 37°C, cells were grown for 24 hours with gefitinib (0, 0.01, 0.1 and 1 μM). The cell number was subsequently obtained using a Cell Counting Kit (Dojindo Laboratories, Kumamoto, Japan) (29). Ten microliters of WST-8 was added to these cells, which were then incubated for 2 hours at 37°C. Optical densities (OD; 450 nm) were obtained with a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA). The status of cell proliferation (%) was calculated according to the following equation: (cell OD value after test materials treated/vehicle control cell OD value) × 100.
Statistical Analysis
Correlations between the clinicopathological factors in the cases examined were evaluated using the chi-square test or Wilcoxon test as appropriate. Student's t-test and the Steel-Dwass test were used for assessing the expression and proliferation in the cell lines. To estimate time-to-event outcomes (OS and PFS), survival curves were determined by the Kaplan-Meier method. Survival differences between groups (high vs. low) were compared by the log-rank test. OS corresponded to the period from the date of surgery to the date of either demise or last contact with the patient, and PFS to that from the date at the start of EGFR-TKI treatment to the date when disease progression was first clinically detected or the last contact with the patient. Both OS and PFS were censored at 5 years after the initial surgery. The statistical analyses were performed using JMP Pro Version 9.0.2 (SAS Institute, Cary, NC, USA). A p value of <0.05 was considered significant in this study.
Results
AGR2 immunoreactivity in lung adenocarcinoma cases
AGR2 immunoreactivity was detected in the cytoplasm of carcinoma cells and adjacent nonpathological alveolar epithelial cells, both type I and II (Fig. 1). Of the lung adenocarcinoma cases, 132/147 (89.8%) were immunohistochemically positive for AGR2. The correlation between clinicopathological factors and the status of AGR2 in the 147 lung adenocarcinoma cases examined are summarized in Table I. The status of AGR2 immunoreactivity in carcinoma cases was significantly associated with that of EGFR gene mutation (p = 0.0013) – i.e., all EGFR gene mutation cases examined were immunohistochemically positive for AGR2. There were no statistically significant associations between the AGR2 status and other clinicopathological factors, including age, sex, tumor size, lymph node metastasis, pathological stage, Ki-67 LI and smoking status.
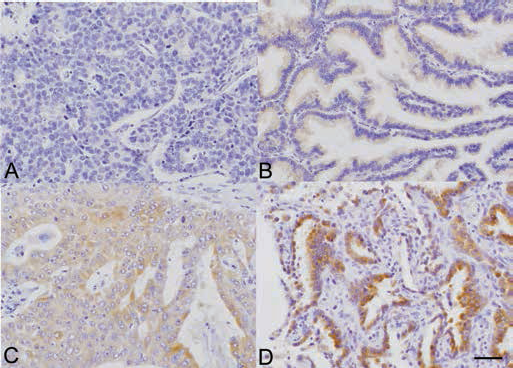
Representative illustrations of anterior gradient 2 (AGR2) immunoreactivity in the cytoplasm of carcinoma cells. These panels demonstrate differences in AGR2 immunostaining intensity in the cytoplasm of carcinoma cells, including 0 = negative (A); 1 = weak (B); 2 = moderate (C) and 3 = strong (D). Scale bar = 50 μm.
Association between AGR2 Immunoreactivity and Clinicopathological Factors
AGR2 IRS was 5.42 ± 3.65 in the 52 EGFR wild cases and 8.73 ± 3.04 in the 49 EGFR gene mutation cases. AGR2 IRS was significantly higher in the EGFR gene mutation cases than in those without mutation (p<0.0001) (Fig. 2A). There were also no significant differences in AGR2 IRS between clinical responders (partial response [PR] and complete response [CR]) and nonresponders (stable disease [SD] and progressive disease [PD]) to EGFR-TKI (Fig. 2B).
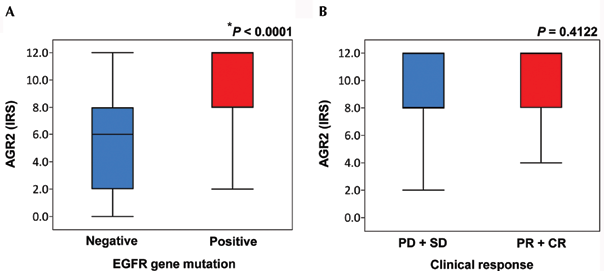
A) Boxplots of anterior gradient 2 immunoreactive score (AGR2 IRS) according to EGFR gene mutation status. AGR2 IRS was 5.42 ± 3.65 (n = 52) among EGFR gene mutation-negative patients, 8.73 ± 3.04 (n = 49) among EGFR gene mutation-positive patients. AGR2 IRS was significantly higher in EGFR gene-mutation patients than in wild-type patients (*p<0.0001, Wilcoxon test). B) Boxplot of AGR2 IRS according to the clinical response to EGFR-TKI. IRS was 8.54 ± 3.35 (n = 11) among nonresponders (stable disease [SD] and progressive disease [PD]) to epidermal growth factor receptor–tyrosine kinase inhibitor (EGFR-TKI) group, 9.47 ± 3.20 (n = 23) among responders (partial response [PR] and complete response [CR]) group.
In this study, we analyzed OS by employing several different levels of cutoff points for the AGR2 IRS (IRS 4/6, 6/8, 8/12). Of the patients, 146 were eligible for this particular analysis. The 5-year OS rates of the AGR2 low IRS groups were 72.82%, 71.7% and 70.15%, whereas those in the AGR2 high IRS groups were 66.49%, 66.47% and 65.04%, respectively, for the different cutoff points (Fig. 3A). Among 49 EGFR gene mutation patients, the 5-year OS rates of the AGR2 low IRS groups were 70.00%, 74.07% and 66.39%, whereas those of the AGR2 high IRS groups were 56.44%, 54.79% and 50.60%, respectively, for the different cutoff levels (Fig. 3B). There were no significant associations between OS and AGR2 IRS at any of the different cutoff levels of IRS. We also evaluated PFS in the same fashion as OS in 34 patients treated with EGFR-TKI. Of these, 33 were eligible in this study, and the median PFS of the AGR2 low IRS groups were 472.5 days, 511 days and 376.5 days, whereas those of the AGR2 high IRS groups were 328 days, 324.5 days and 428 days, respectively, for the different cutoff levels (Fig. 3C). There were no significant associations between PFS and AGR2 IRS at any of the levels examined. These findings all indicated that the AGR2 status was not significantly correlated with OS or PFS, including in those patients treated with EGFR-TKI.
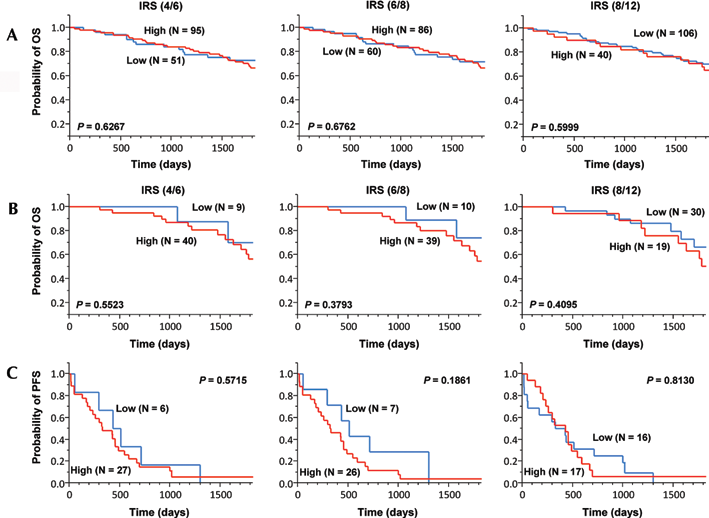
A) Summary of 5-year overall survival (OS) in lung adenocarcinoma patients according to each cutoff point of anterior gradient 2 immunoreactive score (AGR2 IRS; n = 146). There was no significant difference between 5-year OS rate and AGR2 IRS at any cutoff levels of IRS (IRS 4/6, p = 0.6267; 6/8, p = 0.6762; 8/12, p = 0.5999). B) Summary of 5-year overall survival (OS) in lung adenocarcinoma patients harboring EGFR gene mutation according to each cutoff points of AGR2 IRS (n = 49). There was no significant difference between 5-year OS rate and AGR2 IRS at any cutoff levels of IRS (IRS 4/6, p = 0.5523; 6/8, p = 0.3793; 8/12, p = 0.4095). C) Summary of the progression-free survival (PFS) of the lung adenocarcinoma patients harboring EGFR gene mutations according to each AGR2 IRS cutoff point (n = 33). There was no significant difference between PFS and AGR2 IRS at any cutoff levels of IRS (IRS 4/6, p = 0.5715; 6/8, p = 0.1861; 8/12, p = 0.8130).
Effects of AGR2 Expression on Response to EGFR-TKI in Lung Carcinoma Cell Lines
We first evaluated the level of AGR2 expression in the lung adenocarcinoma cell lines. The AGR2 mRNA levels were examined by qRT-PCR (Fig. 4A). The levels of AGR2 mRNA expression in LCSC#1 and PC-3 (EGFR gene mutated) were significantly higher than that in A549 (EGFR gene wild type) (p = 0.0141). Among the EGFR gene mutation cell lines, the level of AGR2 mRNA expression was higher in PC-3 than in LCSC#1 (p = 0.0141). Immunoblotting analysis (Fig. 4B) also revealed a relatively high abundance of AGR2 immunoreactivity in both PC-3 and LCSC#1 compared with A549.
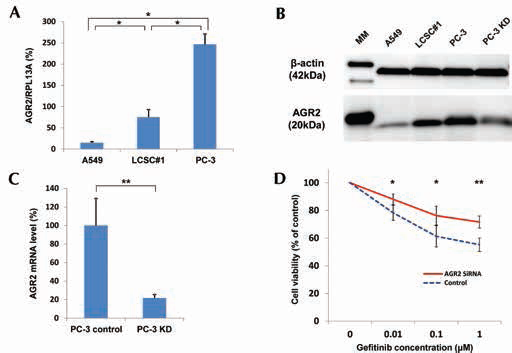
Effects of anterior gradient 2 (AGR2) expression on human lung carcinoma cell lines. A) The levels of AGR2 mRNA expression among the lung carcinoma cell lines, mean ± SD (n = 6). AGR2 expression was significantly higher in LCSC#1 and PC-3 (cell lines with EGFR gene mutations) than in A549 (cell line with EGFR gene wild type; *p = 0.0141, Steel-Dwass test). B) Immunoblotting of AGR2 in A549, LCSC#1, PC-3 and PC-3 transfected with specific AGR2 siRNA. MM = molecular marker. C) The levels of AGR2 mRNA in PC-3 with specific AGR2 siRNA (i.e., PC-3 KD) or control siRNA (i.e., PC-3 control), mean ± SD (n = 3). AGR2 mRNA significantly decreased following the transfection with siRNA (**p = 0.0101, Student's t-test). D) Following the transfection with specific AGR2 siRNA, the response to gefitinib in PC-3 was significantly decreased compared with that in control, mean ± SD (n = 10; *p = 0.0002 and **p<0.0001, Student's t-test).
We then examined the effects of AGR2 expression on the anti-cell proliferative effects induced by gefitinib treatment in PC-3 cells. Both the protein and mRNA transcript levels of AGR2 were significantly decreased following the transfection of specific siRNA against AGR2 (Fig. 4B, C). The cell viability of PC-3 after 24-hour incubation with gefitinib is summarized in Fig. 4D. The cell numbers of PC-3 were not significantly different between AGR2 siRNA and control siRNA transfection. The anti-cell proliferative effects of gefitinib at the concentrations of 0.01, 0.1 and 1 μM were significantly reduced in PC-3 cells transfected with AGR2 siRNA (p = 0.0002, p = 0.0002 and p<0.0001, respectively).
Discussion
In this study, we first demonstrated that AGR2 immunoreactivity was significantly higher in lung adenocarcinoma patients with EGFR gene mutations than in those with wild-type EGFR. AGR2 was initially reported in Xenopus laevis as a homologue of cement gland gene Xenopus Anterior Gradient 2 (XAG2). AGR2 was demonstrated to play an important role in ectodermal patterning, and its overexpression induced the expression of anterior neural marker genes in fibroblast growth factor (FGF) (30). Of interest, in normal human tissues, AGR2 expression was demonstrated in various endoderm-derived organs that contain both mucous-secreting and endocrine cells (31). AGR2 overexpression was reported in human adenocarcinomas derived from the prostate, breast, pancreas and liver (15, 16, 32, 33).
AGR2 immunoreactivity was also reported in surgically resected NSCLC; 85% to 100% of lung adenocarcinoma cases demonstrated AGR2 immunoreactivity in at least >10% of carcinoma cells, and significantly more in lung adenocarcinoma than in squamous cell carcinoma (17, 18). In our present study, AGR2 immunoreactivity was detected in 89.8% (132/147) of the cases examined. Results of in vitro study also demonstrated that AGR2 overexpression in carcinoma cells promoted cell growth, migration and transformation (14), indicating the importance of studying the clinicopathological significance of AGR2 in human lung adenocarcinoma cases. However, it is also true that the methods for evaluating AGR2 immunoreactivity have not been sufficiently established. In this study, we employed the IRS system for the analysis of immunoreactivity because AGR2 was immunolocalized in the cytoplasm, and the IRS system is considered the best available method for the semiquantitative analysis of cytoplasmic immunoreactivity (25). However, the results of our present study did reveal that AGR2 status in adenocarcinoma cells was by no means correlated with the OS or PFS of the patients examined. Therefore, further investigations are required to clarify the roles of AGR2 in the biological behavior of lung adenocarcinoma.
Thirty-four of forty-nine (69.4%) EGFR gene mutation patients were treated with EGFR-TKI. In these 34 patients, the clinical response rate with EGFR-TKI was 67.6% (23/34), and the median PFS was 425 days. The PFS in these patients was significantly longer than that in previous phase III studies (4, 7). However, it is also important to note that this study was limited by the fact that it was a retrospective cohort analysis targeted at patients who underwent surgical resection. Therefore, these patients received a regular follow-up examination following the surgery, and EGFR-TKI was not administered at advanced stages in these patients, which is different from previously reported studies.
The results of our present study in clinical cases at least indicated a very significant correlation between AGR2 overexpression and the EGFR gene mutation status of the adenocarcinoma cells. Our in vitro study also demonstrated that the levels of AGR2 mRNA and protein expression were significantly higher in EGFR gene mutation lung adenocarcinoma cell lines than in those without such mutations. In addition, AGR2 overexpression was significantly correlated with a better therapeutic response to EGFR-TKI in PC-3 harboring EGFR gene mutations. It has thus become important to evaluate the mechanisms underlying AGR2 overexpression in EGFR gene mutation lung adenocarcinoma, and especially to determine whether its overexpression occurs in carcinoma cells as a result of EGFR mutations. AGR2 was recently reported to induce the expression of amphiregulin through the Hippo signaling pathway in adenocarcinoma cells (34). Amphiregulin is one of the well-established ligands for EGFR and promotes cell growth (35, 36). The phosphorylation of the tyrosine kinase following the binding of the ligands to the extracellular domain of the receptor induces cell division, migration, adhesion, differentiation and resistance to apoptosis in carcinoma cells through downstream signaling pathways (37, 38). In lung adenocarcinoma cells, EGFR gene mutations have been reported to be “oncogenic driver mutations,” which dominantly contribute to tumor progression via the constitutively activated signaling pathways (3, 39). EGFR-TKI has also been demonstrated to exert its antitumor effects through competing with adenosine triphosphate (ATP) in the tyrosine kinase domain of the receptor in carcinoma cells. Therefore, AGR2 overexpression is reasonably postulated to increase amphiregulin production in these EGFR gene mutation lung adenocarcinoma cells, which subsequently augment the receptor-mediated effects on carcinoma cells. In the human breast carcinoma cell line, MDA-MB-231, extracellular signal-regulated kinase (ERK) 1/2 inhibitor was recently reported to decrease AGR2 mRNA expression (40). ERK1/2 is also well known to be activated via the EGFR signal (41). These findings all suggest that AGR2 expression reflects the activation of the EGFR signaling pathway in lung adenocarcinoma cells, possibly through the mechanisms described above. In addition to amphiregulin expression, cyclin D1 protein was down-regulated by AGR2 knockdown in both prostate and breast carcinoma cell lines (42, 43). In lung adenocarcinoma cells, cyclin D1 protein (44)/mRNA (45) expression was reported to be significantly higher in lung carcinoma cells with EGFR gene mutations than in those with wild-type EGFR. The reduction of cyclin D1 has also been proposed as a surrogate marker of EGFR-TKI response to NSCLC in both clinical (46) and in vitro (44) studies. Therefore, AGR2 could also be related to the EGFR-TKI response through the activation of cyclin D1 in NSCLC cells. Further investigations are required for clarification of the exact involvement of AGR2 in the biological functions of EGFR gene mutation lung adenocarcinoma cells.
Footnotes
Acknowledgements
We sincerely appreciate the help of Katsuhiko Ono (Department of Pathology, Tohoku University School of Medicine) for scientific assistance.
Financial support: Funding was received from a Grant-in-Aid for Young Scientists (B) and Grant-in-Aid for Scientific Research (B), from Ministry of Education, Culture, Sports, Science and Technology (MEXT), Tokyo, Japan.
Conflict of interest: The authors (M.S., K.M. and H.Y.-O.) have an ownership interest in CHUGAI Pharmaceutical Company, Shizuoka, Japan. The authors (Y.M. and H.S.) have received educational research funding from CHUGAI Pharmaceutical Company. M.S., K.M. and H.Y.-O. are employees of CHUGAI Pharmaceutical Company.