
Abstract
Matrix metalloproteinases (MMPs) are endopeptidases that are involved in extracellular matrix degradation. They are also implicated in a number of abnormal bioprocesses, such as tumor growth, invasion, and metastasis. Therefore, controlling MMP activities has generated considerable interest as a possible therapeutic target. The tissue inhibitors of metalloproteinases (TIMPs) are the major naturally occurring proteins that specifically inhibit MMPs and assist in maintaining the balance between extracellular matrix destruction and formation. However, TIMPs are probably not suitable for pharmacological applications due to their short half-lives in vivo. During the last few decades, synthetic MMP inhibitors (MMPIs) have undergone rapid clinical development in attempts to control MMP enzymatic activities in abnormal bioprocesses. Although studies with these agents have met with limited clinical success, the field of MMPIs is still expanding, and generation of highly effective and selective MMPIs might be a promising direction of this research area.
Introduction
The matrix metalloproteinases (MMPs) are a family of proteolytic enzymes that are involved in catabolizing various components of the extracellular matrix (ECM) (1). Under physiological conditions, the activities of MMPs in processes such as wound healing, morphogenesis, and angiogenesis, are precisely regulated at the transcriptional level as well as post-secretion level, such as zymogen activation and inhibition by endogenous inhibitors. Disrupting the balance between MMPs and their inhibitors may induce pathological conditions associated with uncontrolled ECM turnover, inflammation, cell growth and migration, like those characterizing rheumatoid arthritis, cardiovascular diseases, neurological disorders, and cancer (2-4). Moreover, certain MMPs play contradictory roles at different stages of cancer progression, and thus, have been considered as promising diagnostic and prognostic biomarkers in many types of cancer (5).
During the 1980s, MMPs were first proposed as therapeutic targets for cancer, based on the observations that the metastatic potential of various cancers correlated with the cancer cells’ ability to enzymatically degrade the basement membrane-type collagen (6). Hence, an increasing number of MMP inhibitors (MMPIs) have been developed and evaluated in clinical trials. Because broad-scale MMP inhibition can have both advantageous and problematic effects, MMPI designing over the past decades has focused on the transition from broadly inhibitory hydroxamates to subtype-selective inhibitors.
This review provides an overview of the basic structures of MMPs and the biological functions of their natural inhibitors. We then summarize the history of the development of MMPIs and the related clinical trials. The references listed in the main text contain the most relevant information. As an overview, we tried to summarize all the clinical trials that have been reported during the past decades.
Structure and Members of the MMP family
Structure
The basic structure of MMPs includes an N-terminal signal peptide, a prodomain, a catalytic domain, a hinge region, and a hemopexin domain (Fig. 1). The prodomain, comprising approximately 80 amino acids, contains the cysteine switch motif PRCGXPD. The catalytic domain, comprising approximately 160–170 amino acids, contains a conserved HExxHxxGxxH catalytic zinc binding motif and a binding site for specific substrates. The gelatinases MMP-2 and 9 contain 3 fibronectin repeats within their catalytic domains. The 2 matrilysins MMP-7 and 26 are the smallest MMPs in size, and lack the hemopexin domain. Other MMPs contain a furin-cleavage site that is located in front of the catalytic domain; this site intracellularly activates a Zn2+ zymogen formed by furin. MMP-17 and 25 are inserted into the plasma membrane by a GPI anchor. The membrane-type MMPs (MT-MMPs) are inserted by a 20 amino acid transmembrane domain and contain also a small cytosolic domain. This cytosolic domain may interact with clathrin cages and play an important role in clathrin-dependent internalization of MT-MMPs (7, 8).
Structure of MMPs. SP, signal peptide; H, hemopexin domain; Fn, fibronectin repeat; Fr, furin-cleavage site; TM, transmembrane domain; CS, cytosolic; SA, (N)-terminal signal anchor; Cys, cysteine array; GPI, glycosylphosphatidylinositol (GPI) anchor.
Members of the MMP family
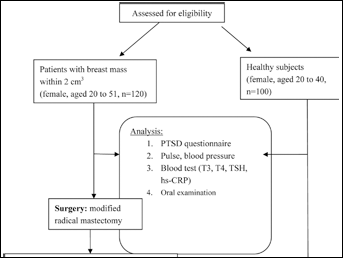
To date, 26 members of the MMP family have been identified in vertebrates; of these, 23 are found in humans. MMPs are divided into 6 groups based largely on their substrates or cellular locations: collagenases, gelatinases, stromelysins, matrilysins, MT-MMPs, and other MMPs (8-10). The chromosomal locations and the substrates for the human MMPs are listed in Table I.
Human Matrix Metalloproteinases
Naturally Occurring MMPIs
TIMPs
TIMP-1 was originally discovered to be a collagenase inhibitor in the culture medium of human skin fibroblasts (11), in human serum (12), and in bovine cartilage and aorta extracts (13). Later, the discovery that it inhibited collagenases, gelatinases, and a proteoglycanase (MMP-3) was the basis for the name “tissue inhibitor of metalloproteinases” or “TIMP” (14).
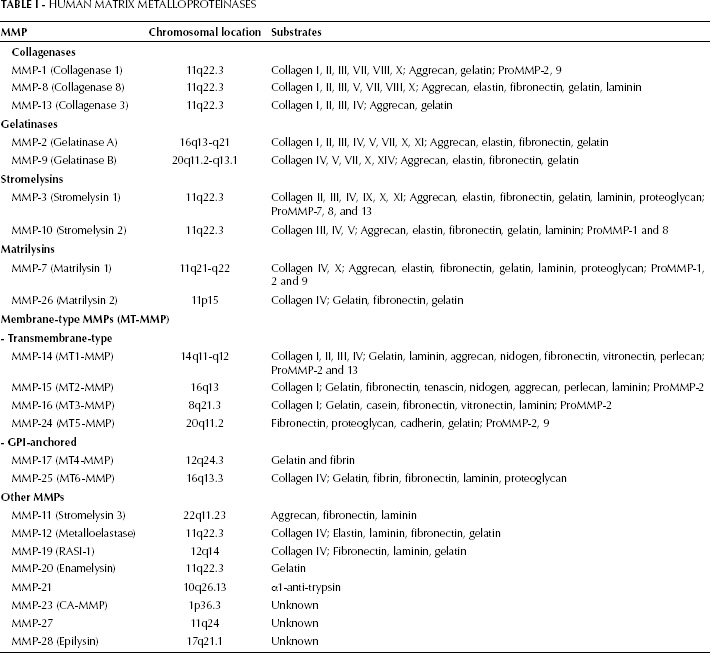
TIMPs are specific endogenous inhibitors that non-covalently bind to MMPs as 1:1 enzyme-inhibitor complexes (15). Four TIMPs (TIMP-1, 2, 3, and 4) have been identified in vertebrates, and their expression is regulated during development and tissue remodeling (16). Some of the general characteristics of TIMPs are summarized in Table II.
General Characteristics Of Human Timps
Fb, fibroblasts; EC, endothelial cells; VSMC, vascular smooth muscle cells.
TIMP functions
In general, the 4 human TIMPs are broad-spectrum inhibitors of the 23 MMPs found in humans, except for TIMP-1 that has a relatively low affinity for the MT-MMPs (17). TIMP-3 has the broadest inhibition spectrum as it inhibits several members of the ADAM (a disintegrin and metalloproteinase) and ADAMTS (ADAM with thrombospondin motifs) families (18). In addition, TIMP-3 binds tightly to the extracellular matrix components (19). Other functions of TIMPs include activating the prometalloproteinases and modulating the extracellular matrix proteolysis, particularly during tissue remodeling and inflammatory processes. TIMPs exhibit growth factor-like activities in certain cell types and inhibit the tumorigenic and metastatic phenotypes of cancer cells (20). TIMP expression is increased by certain cytokines (interleukins-1, 6, and 11, leukemia inhibitory factor, oncostatin M), some growth factors (e.g. transforming growth factor beta), hormones, and retinoids (21, 22).
The balance between MMPs and TIMPs is critical for ECM remodeling. The disruption of this balance can initiate pathological processes in different diseases, such as rheumatoid arthritis, neurological disorders, cardiovascular diseases, and cancer (2-4). Animal studies have shown that TIMP-3 deficiency disrupts matrix homeostasis and causes spontaneous left ventricular dilation and contractile dysfunction, this effect is also associated with elevated MMP-9 activity (23). Another example is that overexpression of MMP-3 in normal epithelium has resulted in spontaneous breast cancers in mice, while overproduction of TIMP-1 has been associated with fewer cancers (4).
TIMP genes and protein structures
The 4 TIMPs have been extensively characterized in terms of their structures, activities, and biological functions. TIMPs are widely distributed among invertebrates and vertebrates, and are somewhat homologous among these organisms (24).
TIMPs with relative molecular mass (Mr) ranging from 21 to 29 kDa consists of 2 distinct structural and functional domains: an N-terminal domain (125 aa) and a C-terminal domain (65 aa). Each domain is stabilized by 3 disulfide bonds (25). TIMP-1 is a 28.5 kDa glycoprotein. TIMP-2 is non-glycosylated and has a Mr of 21 kDa (26). The Mr of TIMP-3 is intermediate between TIMP-1 and 2 (24-25 kDa). TIMP-4 has a Mr of 23 kDa and has been identified as the major MMPI in human platelets (27). The 4 human TIMPs have approximately 40%-50% sequence identity with each other. TIMP-2 and 4 are the most similar, with sequences that are for the 50% identical, whereas TIMP-1 is only 37%-41% identical to the other TIMPs (15). The N-terminal domain of TIMPs (N-TIMPs) has been widely used in characterizing the biochemical and biophysical properties of TIMPs and investigating their structure-function relationships. N-TIMPs function as separate units. They have stable native structures and are fully active inhibitors of MMPs, some ADAMs, and ADAMTSs. The C-terminal domain has at least 2 separate enzyme-binding sites, one for gelatinase A and the other for stromelysin 1 (28-30).
The three-dimensional structures of the full-length TIMP-1, 2, and N-TIMP-3 have been determined by X-ray crystallography (31-36). The solution structures of N-TIMP-1 and N-TIMP-2 have been elucidated by NMR (37-39). All of these structures have a “wedge-shaped” appearance (40).
Alpha-2-macroglobulin (α2M)
α2M is a 718 kDa glycoprotein that is produced by the liver and is found at high concentrations in human plasma. α2M is well known for its broad-spectrum proteinase inhibition ability. The inhibition ability of α2M is achieved in a “trapping” manner. Cleavage of the bait region by a proteinase results in a conformational change in α2M, which then “traps” the proteinase. This process may block the proteinase's access to protein substrates (41). In solution, MMP-1 reacts with α2M more readily than with TIMP-1 (42). However, the large size of α2M may restrict its penetration into tissues and, to some extent, limit its inhibitory effect.
Others
Tissue factor pathway inhibitor-2 (TFPI-2) is a member of the Kunitz-type serine proteinase inhibitor family that inhibits MMPs (43, 44). A C-terminal fragment of the procollagen C-terminal proteinase enhancer protein (CT-PCPE) and its secreted form, membrane-bound β-amyloid precursor protein, inhibit MMP-2 (45, 46). The reversion-inducing cysteine-rich protein with Kazal motifs (RECK) is a GPI-anchored glycoprotein. RECK can suppress MMP-2 and 9 activities, which inhibits angiogenesis. RECK also inhibits the proteolytic activity of MT1-MMP (MMP14) (47, 48). Chlorotoxin, a scorpion toxin that has anti-invasive effects on glioma cells, inhibits MMP-2 (49).
Synthetic MMPIs
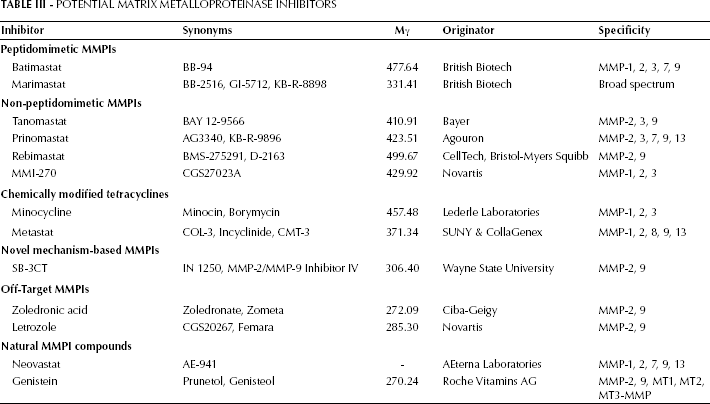
During the last few decades, a considerable number of synthetic MMPIs have been developed to control the synthesis, secretion, activation, and enzymatic activity of MMPs. Several generations of synthetic MMPIs were developed, including peptidomimetics, non-peptidomimetics inhibitors, and tetracycline derivatives (Tab. III) (50). Antisense strategies and small interfering RNA (siRNA) technology can be used to downregulate MMP synthesis by selectively acting against or interfering with the mRNA of a specific MMP (51-53). In addition, various natural compounds that can inhibit MMPs, such as neovastat, a derivative of shark cartilage extract, and genistein, one of several known isoflavones, have been identified (54). Information on clinical trials for these MMPIs is summarized in Tab. IV.
Potential Matrix Metalloproteinase Inhibitors
Mmpi Drugs In Clinical Development
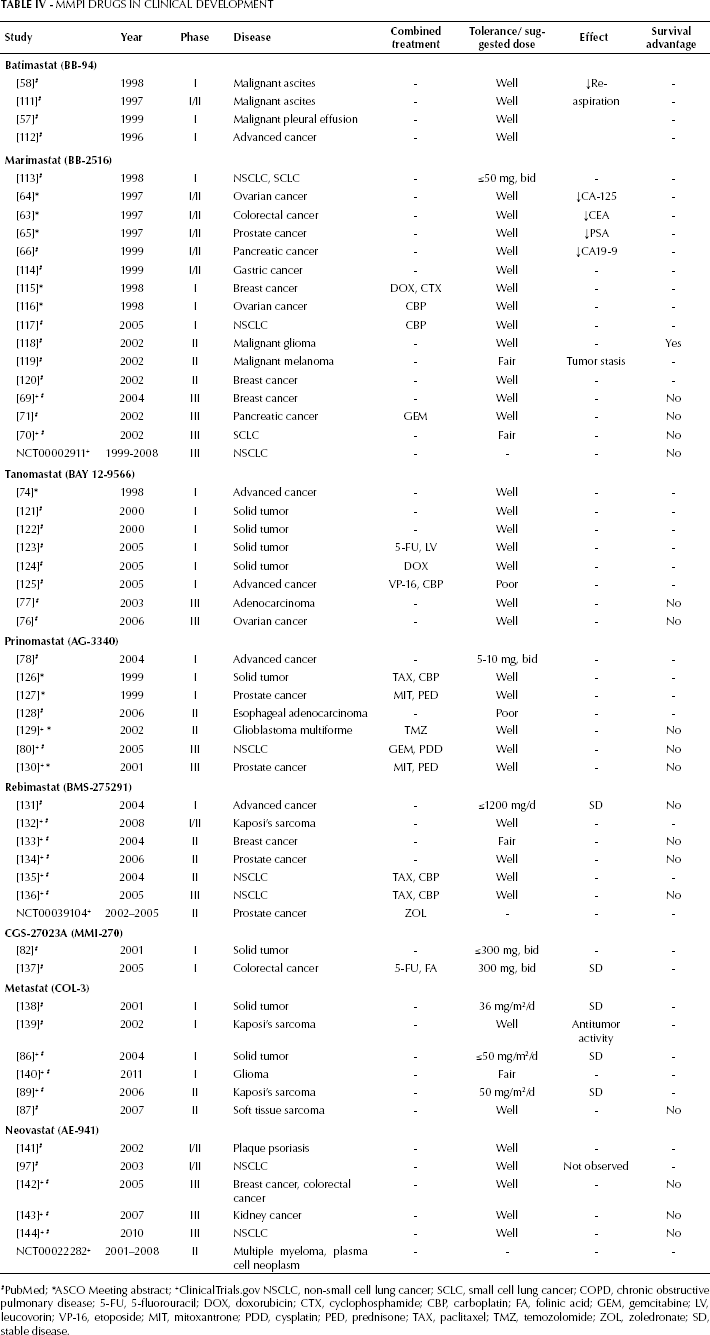
PubMed;
ASCO Meeting abstract;
ClinicalTrials.gov NSCLC, non-small cell lung cancer; SCLC, small cell lung cancer; COPD, chronic obstructive pulmonary disease; 5-FU, 5-fluorouracil; DOX, doxorubicin; CTX, cyclophosphamide; CBP, carboplatin; FA, folinic acid; GEM, gemcitabine; LV, leucovorin; VP-16, etoposide; MIT, mitoxantrone; PDD, cysplatin; PED, prednisone; TAX, paclitaxel; TMZ, temozolomide; ZOL, zoledronate; SD, stable disease.
Peptidomimetic MMPIs
Peptidomimetic MMPIs were synthesized based on a peptide sequence that can be recognized by the targeted metalloproteinase and interacts with the zinc ion of the MMP active site. Based on the chemical structures of the zinc-binding groups, peptidomimetic MMPIs are subdivided into hydroxamates, carboxylates, hydro-carboxylates, sulfhydryls, and phosphoric acid derivatives. The hydroxamate-based compounds are generally considered to be the most potent inhibitors (55).
Phase I/II trials indicated that marimastat had a significant effect on reducing the rising rate of tumor markers in a variety of malignancies in a dose-dependent manner, including colorectal cancer (CEA), pancreatic cancer (CA 19-9), ovarian cancer (CA-125), and prostate cancer (PSA) (62-66). Notably, patients who had these reduced biomarker expressions tended to survive longer than those who did not (67). The most common toxicity associated with marimastat was dose-related musculoskeletal pain and stiffness that may be associated with its broad-spectrum activity. This toxicity was reversible with a one to three-week drug “holiday” being sufficient for resolution, and thereafter most patients were able to continue the treatment with lower dosages (68, 69).
Phase III trials were initiated among patients with metastatic breast cancer, advanced pancreatic cancer, and small-cell and non-small-cell lung cancer. However, these results showed that marimastat did not have superior efficacy in terms of patient survival when compared with either standard chemotherapy or placebo (69-71).
Non-peptidomimetic MMPIs
Non-peptidomimetic MMPIs are designed based on the three-dimensional structures of the MMP active sites. Tanomastat (BAY12-9566), prinomastat (AG3340), rebimastat (BMS-275291), and CGS27023A (MMI-270) are included in this generation (72).
Phase III trials have also been initiated for patients with ovarian cancer and advanced pancreatic adenocarcinoma. Although tanomastat was generally well tolerated, there was no evidence of its effect on progression-free survival or overall survival for ovarian cancer patients (76). In a trial for advanced pancreatic cancer, gemcitabine was shown to be significantly superior to tanomastat (77).
Phase I trials of prinomastat recommended a dose of 5-10 mg twice daily for further studies (78). The primary toxicities were dose and time-dependent joint and muscle-related symptoms, including arthralgia, stiffness, and swelling (79).
The combination of prinomastat, mitoxantrone, and prednisone has been investigated in a phase III trial among hormone-refractory prostate cancer patients. Prinomastat plus gemcitabine and cisplatin for treating patients with advanced non-small cell lung cancer was also investigated in a phase III trial. The primary endpoints in both trials were overall survival and the time to tumor progression. However, results for both trials failed to show that prinomastat has the potency to improve the clinical outcomes of chemotherapy for these patients (80).
Phase I/II trials suggested that 1,200 mg once daily was a tolerated dose. Joint and muscle toxicities were not dose-related when using rebimastat. Other reported toxicities were rash, fatigue, headache, nausea, and changes in taste.
Rebimastat plus paclitaxel and carboplatin went through a phase III trial for patients with advanced non-small cell lung cancer. The result of this study was disappointing as the combination increased the toxicity of rebimastat and no improvements in survival were observed.
Chemically modified tetracyclines
Using the strategy of blocking gene transcription, tetracycline derivatives can inhibit both the enzymatic activity and synthesis of MMPs (83). Compared with regular tetracyclines, chemically modified tetracyclines do not have antibiotic activities and cause limited systemic toxicity. They may inhibit MMPs by binding to metal ions, such as zinc and calcium. Metastat (COL-3), minocycline, and doxycycline belong to this family of MMPIs. Doxycycline, a chemically modified tetracycline, is currently the only MMPI approved by the Food and Drug Administration for use as an inhibitor of MMP-7 and 8, which are involved in periodontitis (84).
Novel mechanism-based inhibitors
SB-3CT (MMP-2/MMP-9 inhibitor IV) is a potent, selective, slow binding, and the first competitive mechanism-based inhibitor of human gelatinases. The fundamental step involved in its inhibitory mechanism is an enzyme-catalyzed ring opening of thiirane, which results in a stable zinc-thiolate species (90). Studies using mouse models reported that it could suppress liver metastasis and increase animal survival (91).
Off-target MMPIs
Off-target MMPIs refer to certain drugs that have already been shown to have beneficial effects on MMPs and other ECM molecules that were not their intended primary targets.
Examples of this are the bisphosphonates that inhibit the function of the mevalonate pathway. In addition to their inhibitory effects on osteoclast activity and bone resorption, bisphosphonates inhibit the enzymatic activities of various MMPs (92). Moreover, one in vitro study showed that zoledronic acid, a third generation bisphosphonate used as a standard treatment for preventing skeletal complications associated with bone metastases, was particularly associated with downregulation of MMP-2 and 9 expression (93).
Another off-target MMPI is letrozole, a reversible nonsteroidal inhibitor of the P450 aromatase. Letrozole is a type of aromatase inhibitor that is widely used in hormone therapy for breast cancer patients. Studies have shown that letrozole could suppress the release of gelatinases (MMP-2 and 9) by breast cancer cells in a dose-dependent manner (94, 95).
Natural MMPI compounds
Conclusions and Future Prospects
MMPs form well-established complexes and are integrated in the communication networks within tissues and cells for regulating cell proliferation, differentiation, tissue homeostasis, immune responses, and a number of other processes. In addition, they play key roles in cancer progression. However, in most clinical trials, the use of agents that targeted MMPs resulted in poor outcomes, which were not consistent with preclinical studies (102). There are several explanations for these contradictory outcomes.
First, recent studies showed that members of the MMP family had different functions at different stages of cancer development. Certain MMPs may even have protective functions against cancer. In these cases, the use of broad-spectrum MMPIs may result in poor clinical outcomes (103). Second, the toxicities of MMPIs, such as the most common musculoskeletal syndromes, have limited the maximum-tolerated doses of certain drugs, and thus, restricted the drugs’ efficacies.
For these reasons, developing inhibitors that are specific for certain MMPs but do not cross-react with other MMPs is essential for future development of MMPIs (104). One possible strategy for increasing the specificity of MMPIs is using drugs that target specific exosites related to MMP substrate selectivity. Exosites are binding sites outside of the active domains of MMPs (105, 106). Examples are the phosphinate triple-helical transition state analogs that have high affinity and selectivity for MMP-2 and 9 (107). Therefore, future drugs targeting less conserved exosites rather than catalytic domains will be both MMP and substrate-specific. On the other hand, studies have shown that microRNA miR-98 plays a regulatory role in tumor angiogenesis and invasion through repression of MMP-11 expression (108); this observation suggests that microRNAs might also possess therapeutic potential in MMP regulation. Furthermore, MMP expression is usually associated with expression of other molecules in pathological processes. For example, the expression of telomerase reverse transcriptase mRNA in breast cancer is associated with MMP-1 and VEGF expression (109). Another example is that cyclooxygenase 2 (COX-2), MMP-1 and MMP-2 cooperate to mediate primary tumor growth and angiogenesis, as well as pulmonary metastasis in breast cancer (110). These associations suggest that specific drug combinations may be more effective than the use of MMPIs only. In addition to this, clinical trials of MMPIs combinations suggest that an appropriate combination of these compounds with other chemotherapeutic or molecule-targeted agents may be important for increasing drug efficacy.
In conclusion, the major lesson we could get from the clinical trials here considered is that we need to improve the methodology to avoid or minimize the adverse events. As a result, developing a new generation of effective and selective MMPIs is an emerging and promising area of research.