
Abstract
Recent epidemiological studies and animal experiments have demonstrated that nonsteroidal anti-inflammatory drugs (NSAIDs) reduce the incidence of colorectal carcinoma. Cyclooxygenase (COX) is the principal target of NSAIDs. COX is the first oxidase in the process of prostaglandin production from arachidonic acid. COX enzyme may be involved in the initiation and/or the promotion of tumorigenesis due to NSAIDs inhibition of COX. Lipoxygenase (LOX) is also an initial enzyme in the pathway for producing leukotrienes from arachidonic acid. Similar to COX, LOX enzyme may also be involved in the initiation and/or promotion of tumorigenesis. Peroxisome proliferator activator-receptor (PPAR)-γ is a ligand-activated transcriptional factor belonging to the steroid receptor superfamily. PPAR-γ plays a role in both adipocyte differentiation and tumorigenesis. PPAR-γ is one target for cell growth modulation of NSAIDs.
In this review, we report the expression of COX-2, LOX and PPAR-γ in human bladder tumor tissues as well as the effects of COX-2 and LOX inhibitors and PPAR-γ ligand.
Introduction
The natural history of the bladder tumor (BT) is not well understood, but exposure to carcinogens, including aromatic amines, is considered a major risk factors for the development of BT. Workers exposed to aromatic amines frequently have a mutated p53 gene, a tumor suppressor gene involved in the tumorigenesis of many tumors.
Non-steroidal anti-inflammatory drugs (NSAIDs) have anti-tumor effects for the RCC patient, thus, attracting a great deal of attention. The typical target of NSAIDs is cyclooxygenase (COX). In recent reports, a number of patients have had significantly low risks of colorectal cancer while they continued using NSAIDs typified by aspirin. Consequently, the suppression of tumorigenesis by administering NSAIDs has come into focus. It was also reported that the size and number of adenoma were markedly reduced when sulindac, a type of NSAIDs was given to patients with familial adenomatous polyposis, a high risk group for colorectal cancer. 1
It is known that NSAIDs inhibit the activity of COX and production of prostaglandin. NSAIDs also stimulate peroxisome proliferator activator-receptor (PPAR)-γ and inhibit the production of chemical mediators such as tumor necrosis factor-α, interleukin-1β and interleukin-6 through the expression of PPAR-γ in leukocytes. PPAR-γ is thus a promising target for cell growth modulation by NSAIDs.
In this review, we discuss the possibility that the target of arachidonic acid pathway metabolite may be a new anti-tumor strategy for human BT.
Arachidonic Acid Pathway
The metabolism of arachidonic acid by either COX pathway or lipoxygenase (LOX) pathway generates eicosanoids, which have been implicated in the pathogenesis of a variety of human diseases, including cancer, and are considered important in tumor promotion, progression, and metastasis. 2
COX is the first enzyme in the pathway for producing prostaglandin (PG) and thromboxane (Tx) from arachidonic acid, and can occur as three isoforms, COX-1, COX-2 and COX-3. The enzymes of both COX-1 and COX-2 are transformed from the cell membrane phospholipid to arachidonic acid by the phospholipase A2, and then transform arachidonic acid to PGH2 through PGG2. COX-1 occurs in tissues and cells and works to protect the cell. COX-2 express momentarily and strongly in response to growth factors and some endotoxins. It is involved with inflammation, cell proliferation and differentiation. 3 Recently, COX-2 has also been shown to play an important role in tumorigenesis. 1 Although the existence of COX-3 has recently been reported, it continues to be argued. In pathogenesis of BT, Both COX-2 and PGs (especially PGE2) play a role in development of BT.
LOX is the first enzyme in the pathway for producing leukotriene (LT) from arachidonic acid. Isoenzymes of LOX include 5-LOX, 12-LOX and two 15-LOX isoforms (15-LOX-1, 15-LOX-2). These catalyze the biosynthesis of biologically active compounds such as LTs and hydroxyeicosatetraenoic acids (HETEs).4,5 5-LOX catalyzes the first step in oxygenation of arachidonic acid to produce 5-hydroperoxyeicosatetraenoic acid (5-HPETE), and the subsequent metabolism of 5-HPETE to 5-HETE and LTs. LTs belong to an important group of pro-inflammatory mediators that are synthesized from arachidonic acid via the 5-LOX pathway. The activity of 5-LOX leads to the formation of unstable LTA4, which can be converted into either LTB4, or cysteinyl LTs (LTC4, LTD4 and LTE4). 6
The 12-LOX, includes platelet 12-LOX, and leukocyte 12-LOX that oxygenate arachidonic acid at position C-12 to produce 12-hydroperoxyeicosatetra-enoic acid and then 12-HETE. 2 Whereas 5-LOX, 12-LOX and 15-LOX-1, have pro-tumorigenic roles, 15-LOX-2 appears to have an anti-tumorigenic roles. The administration of LOX inhibitor may inhibit matrix metalloproteinase (MMP)-2, it may induce down regulation such as BCL-2 and nuclear factor-kappaB (NF-λB), and it may activate such as PPAR-γ, apoptosis activating factor-1 and caspase-3.
PPAR
PPARs are members of the nuclear receptor super-family of ligand-activated transcriptional factor such as steroids, thyroid hormone, vitamin D3 and retinoic acid. 7 PPAR binds to peroxisome proliferator response element (PPRE) as a heterodimer with the retinoic receptor (RXR) in the regulation of PPAR target genes. PPARs may be important immunomodulatory factors as well as fatty acid regulators. PPARs modulate these activities in different immune cell types such as monocyte/macrophages, lymphocytes, and endothelial cells.
Three PPAR subtypes (α, β, and-γ) have been identified. PPAR-α is highly expressed in the liver, heart, kidney, muscle, brown adipose tissue, and gut tissues which exhibit high carbolic rates towards fatty acid. 8 PPAR-β is expressed ubiquitously, and its function is relatively unknown. 9 Recent studies suggest that PPAR-β may be a target for NSAID-induced tumor suppression in colorectal tumors. PPAR-γ is expressed at high-level in adipose tissue and is a critical regular of adipocyte differentiation. In addition, PPAR-α, and –γ may be important immunomodulatory factors. PPAR-α-knockout mice exhibit exacerbated inflammatory responses, and LTB4, a chemotactic mediator, seems to regulate the clearance of itself as an agonist of PPAR-α. PPAR-γ is also expressed in the immune system tissues and cells (spleen, bone-marrow, monocytes, helper T-cell clones) and skeletal tissues (bone, synovioum, chondrocytes). 8 PPAR-γ also have an anti-tumorigenic roles.
Recent data have shown that PPAR-γ ligands lead to inhibition of phorbol ester-induced nitric oxide and macrophage-derived cytokines. PPAR-γ ligands also induce apoptosis in macrophage, fibroblasts, and endothelial cells. 10 PPAR-γ plays a role in both adipocyte differentiation and tumorigenesis. PPAR-γ ligands lead to inhibition of the expression of nitric oxide, cytokines, chemokines and adhesion molecules, in part by antagonizing the activities of transcriptional factors. Furthermore, PPAR-γ ligands including anti-diabetic thiazolidinedione (such as troglitazone) and 15-deoxy-Δ12,14 -prostaglandin J2 (15-d-PGJ2) have potent tumor modulatory effects on several tumors.11,12
15-d-PGJ2 induces apoptosis in macropharge, endothelial cell, and choriocarcinoma cell13-15 as well as thiazolidinediones-induced fibroblast apoptosis.
16
PPAR-γ ligands also inhibit vascular endothelial cell growth factor-induced angiogenesis
COX and BT
COX-2 expression in BT tissues was stronger than in chronic cystitis (CC) and normal bladder (NB) tissues by immunohistochemical staining (118 BT, 10 CC and 8 NB tissues) or RT-PCR (Fig. 1). We classified 3 categories (epithelium, blood vessel, stromal tissue) in BT tissues, and examined them for intensity of COX-2 expression. COX-2 expression was more intensive and extensive in all categories of BT tissues than CC and NB tissues. Significant differences occurred between grades of BT tissues in only epithelium in COX-2 expression. COX-2 expression was stronger in G3 cancer and advanced-stage BT (pT2 or above). 21

Mohammed et al. reported the expression of COX-1 in both BT and NB tissues by immunohistochemical staining and western blot. COX-2 was stronger in the advanced stage BT than NB tissue. 22 Komhoff et al. reported COX-2 expression was stronger in low differentiation BT by immunohistochemical staining. 23 Shirahara et al. reported COX-2 expression was stronger in the advanced-stage BT, and COX-2 expression was most strongest in the highest malignancy of BT in situ, and COX-1 expression was nothing in any BT tissues by immunoblot and immunohistochemical staining. 24 Margulis et al. reported COX-2 was not expressed in NB urothelium. COX-2 over expression is associated with pathological and molecular features of biologically aggressive disease, suggesting a role for COX-2 in BT development and invasion. 25
Okajima et al. reported the administration of nimesulide and piroxicam as selective COX-2 inhibitor could reduce remarkable lowering of tumorigenesis rate in the BT using rat model which superficial BT initiating the tumor by administering the N-butyl-N-nitrosoamine. 26 Okamoto et al. reported etodolac (selective COX-2 inhibitor) exhibited anti-tumor activity and induced E-cadherin expression in BT cells and might be useful for the clinical treatment and prevention of BT, especially in poorly differentiated bladder cancer with high COX-2 and low E-cadherin expression. 27
However, recently accepted explanation is that only celecoxib, among the COX-2 inhibitors induces apoptosis of cancer cells. We suggest the increased apoptosis cells produced by celecoxib might be associated with decreased PGE2 production.
Generally, Bcl-2 is a key factor suppressing the apoptosis accompanying of caspase. Gee et al. reported treatment with 100 μM celecoxib resulted in significant apoptosis in three kinds of BT cell lines, which was associated with down-regulation of Bcl-2. 28
In our experiment, we could not demonstrate significant apoptosis induction by administering eight kinds of COX-2 inhibitors (10–80 μM) into the human BT cell lines (T24) (Table 1). Additionally, we could not demonstrate the significance of both selective COX-2 inhibitor (etodolac, meloxicam, nimesulide and NS398) and relatively poor selective COX-2 inhibitor (ibuprofen, indomethacin, piroxicam and s-naparoxen). 29
Effects of COX and LOX inhibitors, and PPAR-γ ligand in viability of human BT cell line (T24).

The dose-response analysis of viability in human BT cell line (T24) treated with COX-2 and LOX inhibitors, and PPAR-γ ligand (5–80 μM, 48 hr) was measured by the MTT assay and expressed as % of control culture conditions.
In conclusion, COX-2 expression is strong in BT, but the anti-tumor effect of COX-2 inhibitor is very weak in BT patients in a single administration at a clinical dose. It may be difficult to suppress the growth of BT for chemotherapy. COX-2 inhibitor is suitable for chemopreventive therapy.
LOX and BT
We have shown that 5- and 12-LOX expressions in BT tissues were stronger than those in NB tissues by immunohistochemical staining (170 BT, 20 CC and 20 NB tissues) (Fig. 2) and RT-PCR. We classified 3 categories (epithelium, blood vessel, stromal tissue) in BT, CB and NB tissues, and examined the intensity of 5- and 12-LOX expressions. 5- and 12-LOX expressions were more intensive and extensive in all categories of BT tissues as compared with CC and NB tissues (Table 2). 5-LOX expression was stronger in G3 cancer and advanced-stage BT (pT2 or above) than G1 cancer and early-stage BT (pT1 or below). 2
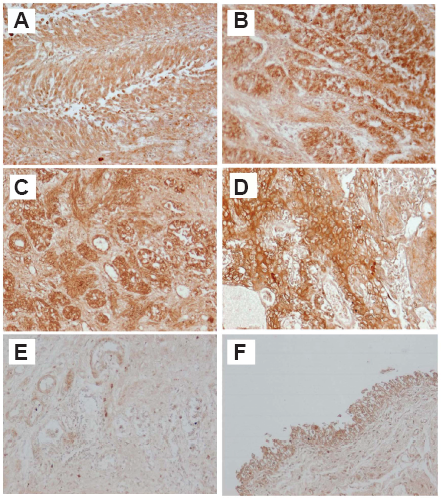
Statistical analysis of 5- and 12-LOX immunostaining.

p < 0.001.
At 10–80 μM, some, but not all LOX inhibitors reduced the viability of BT cell lines (T24) by MTT assay (Table 1). 5-LOX inhibitor appeared more potent than the 12-LOX inhibitor. 30 BT cells treated with some LOX inhibitor (50 μM) showed chromatin condensation, cellular shrinkage, apoptotic bodies, and cytoplasmic condensation by hoechest staining (Fig. 3).

BT cells treated with 5-LOX inhibitor (100 μM) also entered early apoptosis, but not late apoptosis, necrosis or DNA fragmentation by flow cytometry.
Hayashi et al. reported BT cells frequently expressed 5-LOX. 5-LOX inhibitor (AA861) revealed the strongest growth suppression of those cells compared to other LOX and COX pathway inhibitors, and the growth suppression effects were considered to be due to inhibition of the enzymatic activity. 31
In conclusion, 5-LOX expression is strong in BT, especially low differentiation and advanced-stage BT. The anti-tumor effect of 5-LOX inhibitor is significantly stronger than those of COX-2 inhibitor. The anti-tumor effect of 5-LOX inhibitor is weak in BT patients in a single administration at a clinical dose. 5-LOX inhibitor is suitable for chemopreventative therapy, like COX-2 inhibitor.
PPAR-γ and BT
By immunohistochemical staining and RT-PCR, PPAR-γ expression is increased in BT tissues as compared with NB tissues (170 BT and 20 NB tissues). In all categories (epithelium, blood vessel, stromal tissues) of BT and NB tissues, the intensity of PPAR-α, -β expression was not significantly different. In all categories, PPAR-γ expression was significantly greater in BT tissues than in NB tissues. Only epithelium, PPAR-γ expression was stronger in G3 cancer. In all categories, PPAR-γ expression was stronger in advanced-stage BT (pT2 or above). 32
At 10–40 μM, PPAR-γ ligands (troglitazone and 15-d-PGJ2) reduced the viability of BT cell lines (T24) by MTT assay (Table 1). BT cells treated with PPAR-γ ligands (25 μM troglitazone and 15-d-PGJ2) could early apoptosis, not late apoptosis or necrosis and DNA fragmentation by flow cytometry (Fig. 4). PPAR-γ ligands (20 μM troglitazone and 15-d-PGJ2) also induced apoptosis in BT cells by hoechest staining. 33

About anti-tumor mechanism of troglitazone and 15-d-PGJ2, Chaffer et al. reported troglitazone induced G0/G1 growth arrest while 15-d-PGJ2 induced apoptosis. Troglitazone and 15-d-PGJ2 inhibit growth of BT cell lines through different mechanisms and the effects of both agents are PPAR-γ independent. 34
Bacillus Calmette-Guérin (BCG) is considered to be one of the most effective treatments for superficial and in situ BT. Lodillinsky et al. reported BCG induced functional PPAR-γ in BT cells in vivo and in vitro, being these receptors intrinsically involved in the anti-tumor activity of BCG. 35
In conclusion, PPAR-γ is strong in BT, and the anti-tumor effect of PPAR-γ ligand is significantly greater than that of 5-LOX inhibitor. The anti-tumor effect of PPAR-γ ligand is relatively weak. Targeting PPAR-γ in BT is likely to be useful more as a chemopreventive rather than chemotherapeutic.
Conclusions
There is no discussion that COX-2, LOX (especially, 5-LOX) and PPAR-γ are involved in the initiation and promotion of BT tissues. It may be possible to use COX-2 and 5-LOX inhibitor, and PPAR-γ ligand as an anti-tumor drug for chemopreventive therapy in a single administration at a clinical dose. However, it may be difficult to use the COX-2 and 5-LOX inhibitor, even if PPAR-γ ligand in single administration at a clinical dose in expectation for chemotherapeutic therapy. However, the clinical application of PPAR-γ ligand and 5-LOX inhibitor require further research and consideration, the target of PPAR-γ ligand and 5-LOX inhibitor is a novel strategy over human BT.
We conclude the administration of COX-2 and 5-LOX inhibitor, and PPAR-γ ligand are useful with usual treatment in human BT. In near future, the combination therapy of COX-2 and 5-LOX inhibitor, and PPAR-γ ligand will be useful new treatment of human BT.
Disclosure
The authors report no conflicts of interest.