The proteins of an Antarctic bacterium Pseudomonas syringae Lz4W, identified earlier by different membrane protein preparation methods, were combined together and the redundant identities removed. In total, 1479 proteins including 148 outer membrane proteins from this bacterium were predicted by the algorithm PSORTb3.0. A detailed analysis on their subcellular localization was undertaken which was determined using TMHMM, TMB-hunt and BOMP. A comparison of PSORTb predicted outer membrane proteins with BOMP, revealed that most of the proteins predicted by the former, contained β–barrels in the outer membranes. A comparative analysis of PSORTb, TMHMM and TMB-hunt reveals that most of the outer membranes proteins of this bacterium could be identified using this approach. Thus, by using a combination of biochemical and different bioinformatics algorithms, the membrane proteins of P. syringae are analyzed. In particular, PSORTb results are compared and supported by other algorithms, to improve the strength of OM proteins prediction. Several proteins, having an important role in cold adaptation of the organism, could also be identified.
In proteomics, large numbers of proteins are identified by tryptic digestion of proteins followed by a LC-MS/MS analysis.1–4 Pre fractionation and/or separation of proteins by 1D SDS PAGE in combination with LC-MS/MS helps in detecting the low abundant proteins for their identification. All gram-negative bacteria contain a complex outer membrane (OM), which envelopes the cell from outside. The space between the outer membrane and the cytoplasmic membrane (CM) is called periplasmic space, which contain loose network of peptidoglycan chains. Each of the subcellular components contains unique set of proteins. The subcellular localization (SCL) of the proteins is an important indicator of their function in the cell.
The proteins of the Antarctic bacterium Pseudomonas syringae were fractionated using different methodologies and the proteins were identified using conventional methods. The membrane proteins of this bacterium were prepared using several techniques viz, enrichment and purification of outer membrane proteins (OMPs), solubilization of these proteins, their identification and characterization.5,6 Each of these experimental steps is beset with difficulties mainly because of the intractable nature of the hydrophobic membrane proteins in the inner and outer membrane.7–9 Our earlier studies revealed that sucrose density gradient method provides better option for the preparation of OM proteins from the Antarctic bacterium P. syringae.6
Studies on the mechanism of cold adaptation using psychrotrophic bacteria revealed that changes in the structures of membranes and the membrane components play an important role.10–16 The possible role of membrane proteins of the Antarctic bacterium P. syringae in sensing the environmental temperature was studied in our laboratory.17,18 Preparation of membrane proteins without co-purification of non-membrane proteins is an uphill task and several approaches are being developed to enrich the membrane proteins.8,9
The Antarctic bacterium P. syringae is being used as a model organism to study cold adaptation in our laboratory. The membrane proteins were prepared using different protocols such as sucrose density gradient, Triton X-100 solubilization method, and carbonate extraction method. The proteins were separated on 1D and 2D gels, digested with trypsin and then identified using LC-MS/MS and MALDI TOF/TOF analysis. The proteins identified from the mass spectra of the tryptic peptides obtained from LC-MS/MS were identified using SEQUEST, where as the protein digests obtained from 2D gel spots were analyzed by MALDI TOF/TOF and identified using MASCOT. As the genome sequence of this bacterium is not known, the genome sequences of other Pseudomonas sp. already available at NCBI were used to identify the proteins.5,6
It is possible to predict proteomes and protein localizations in silico based on the genomic information using different algorithms developed for this purpose. As the experimental protocols have their own limitations, particularly in case of membrane proteins, different algorithms were also used to identify the subcellular localizations.
In the present study, the biochemical and bioinformatic approaches were suitably combined to delineate the subcellular localization of the membrane proteins. The functional significance of some proteins identified in this study is also discussed.
Methods
Preparation of Membrane Proteins
The membrane proteins of the Antarctic bacterium P. syringae were prepared using Triton X-100 solubilization as described earlier5 (Jagannadham 2008), carbonate extraction procedure20 (Molloy et al 2000) and sucrose density gradient method as described earlier6,21 (Gotoh et al 1994; Jagannadham et al 2011).
Sub Cellular Localization Using Psortb
All the membrane proteins of P. syringae Lz4W obtained using different protocols were listed together. The protein redundancy was removed based on the protein name, accession number and sequence identity.5,6 In all, a total of 1479 proteins were identified. The sub cellular localization of these proteins was predicted using the PSORTb version. 3.0.2 program (http://www.psort.org) by choosing gram-negative long format (tabdelimited). The output displays the final predictions and associated scores as described earlier.22 The cytosolic proteins were clearly predicted with good scores (8.96–10 on a scale of 10). The transmembrane α-helix predictor module HMMTOP23,24 used in the earlier version 2.025 for the prediction is replaced with S-TMHMM in the latest version 3.0 to improve the prediction of helices.22 The predictions made for each protein are shown in the Supplemental Table 1. The protein sequences were submitted in the FASTA format for all the predictions.
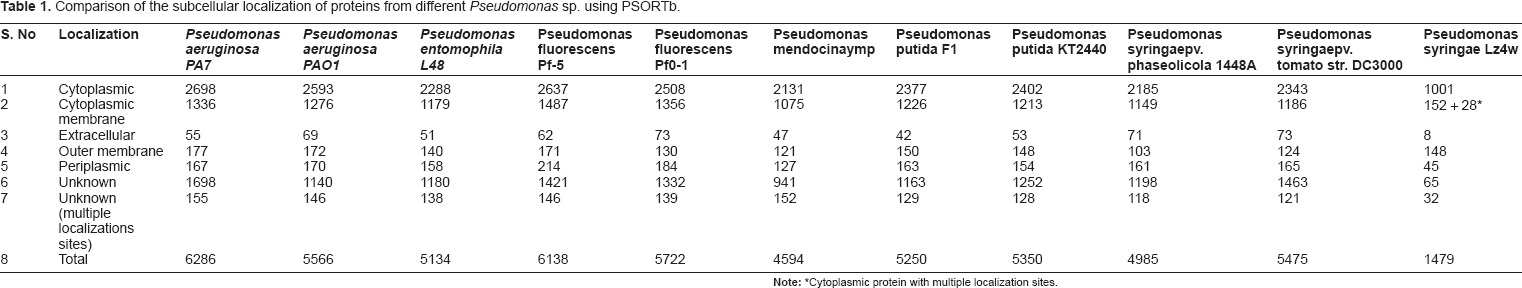
Comparison of the subcellular localization of proteins from different Pseudomonas sp. using PSORTb.
Cytoplasmic protein with multiple localization sites.
Predicting β-Barrels of OMP's
The β-barrel outer membrane predictor (BOMP) calculates and predicts the polypeptide sequences of the gram-negative bacterium for this structural motif. The β-barrel of the integral membrane proteins from the outer membrane proteins of P. syringae Lz4W have been predicted using the program BOMP as described earlier.26 Using this algorithm the sequence predicted for the β-barrel proteins are given scores from 5 to 1. The values obtained for each protein are shown in the Supplemental Table 1. The probability of the protein submitted for prediction being a β-barrel protein increase with increase in the number. This program combines two methods, one for identifying the C-terminal pattern typical of many integral β-barrel proteins27 (Struyve et al 1991), and the other a filtering mechanism to remove the false positives based on sequence stretches that contain amino acids typical of transmembrane β-strands28 (Wimley 2002). This program also has a provision to verify subcellular localization. The proteins that do not contain these domains will be omitted in the computation.
TMB-Hunt
TMB-Hunt discriminates between β-barrel trans membrane (bbtm) domains and non-bbtm containing proteins based on their amino acid composition as described by Garrow29 et al 2005. The membrane proteins of P. syringae Lz4W prepared using different methods were analyzed using this program. The scores for predicting bbtm domains show positive values and non-bbtm proteins show negative values ranging from 1 to -1. Proteins containing bbtm and exhibiting values from 0.2 to 1 are only included in the present predictions.
Predicting Trans-Membrane Helices
The trans-membrane topology of the membrane proteins can be predicted using TMHMM program described by Sonnhammer30 et al 1998. This algorithm can discriminate between soluble and membrane proteins with high specificity and sensitivity. The membrane proteins of P. syringae Lz4W were analyzed using this algorithm, the numbers of trans-membrane helices present in each protein are predicted and shown in Supplemental Table 1.
Results and Discussion
Several membrane protein preparation methods also co-purify proteins from other subcellular locations of bacteria. Hence it is important to localize these proteins using other bioinformatics approaches. This study aims at predicting the subcellular localization of the proteins prepared and identified using different membrane protein preparation methods from an Antarctic bacterium P. syringae Lz4W. Integral membrane proteins from the cytoplasmic membrane and the outer membrane differ in their secondary structure content and amino acid composition. The cytoplasmic membrane proteins are mainly composed of hydrophobic α-helices, whereas outer membrane proteins are mainly composed of β-barrel motifs.31–33 Several algorithms are available to predict the subcellular localization of the proteins based on the physico-chemical and conformational properties. Even though several efficient algorithms are available to predict subcellular localizations of proteins in Gram-negative bacteria, no single algorithm is capable of predicting localizations of all proteins very efficiently. Hence different algorithms have been used to predict the subcellular localizations of the proteins of P. syringae Lz4W. All the predictions made for subcellular localizations, and further prediction of α-helices and β-barrels to distinguish CM and OM proteins respectively using various algorithms are shown in the Supplementary Table 1 against each protein.
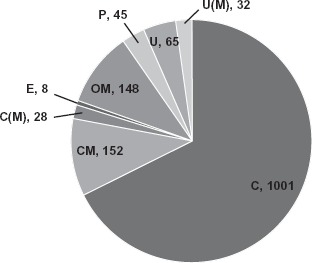
The subcellular localization of the proteins identified from P. syringae Lz4W have been predicted using PSORTb V.3.0.2 as shown in Figure 1. It is clear from these results that out of total 1479 proteins, 148 proteins are from OM and 152 are from CM as predicted by the algorithm (Supplemental Table 1). The location of 65 proteins could not be predicted, whereas 32 proteins predicted unknown localization, contain multiple localization sites. In addition there are 28 cytoplasmic proteins with multiple localization sites. It could also be possible that some these proteins are also part of membrane proteins.
The sub cellular localization of the proteins in P. syringae Lz4W, predicted by PSORTb.V.3. The number of proteins predicted in each subcellular location, OM-outer membrane, CM-cytoplasmic membrane, E-extracellular, P-periplasmic, C-cytosol, C(M)-cytoplasmic with multiple localization sites, U-unknown, U(M)-unknown location with multiple localization sites, is shown.
The results of subcellular localization of different Pseudomonas sp is available at the PSORTb web site. According to these predictions Pseudomonas aeruginosa PA7 contains the highest number of 177 OM proteins, whereas the Pseudomonas syringae pv. phaseolocola 1448A has the lowest number 103 OM proteins. For the other entire Pseudomonas sp, the OM proteins lie in between 103 to 177. From this analysis it is not farfetched to assume that the OM proteins of the other Pseudomonas sp lie in this range. Using the same algorithm the SCL of P. syringae Lz4W predicted 148 OM proteins. Therefore, a comparison for the predictions of the SCL of proteins from the Pseudomonas sp was made with the SCL proteins of P. syringae Lz4W, which revealed that almost all the outer membrane proteins of P. syringae Lz4W were identified (Table 1). Even though PSORTb continues to be the most precise subcellular localization predictor of its kind till date, even the new improved version does not discriminate between integral β-barrel proteins and OM lipoproteins (Gardy et al 2005, Yu et al 2010). Therefore, other prediction algorithms such as BOMP and TMB-hunt were also adopted to predict the integral membrane proteins.
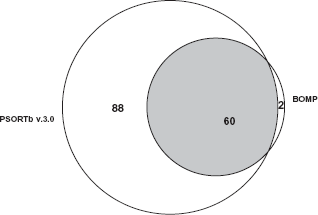
Using BOMP algorithm 62 proteins were predicted to contain β-barrel motifs. Comparison of these results with PSORTb showed that most of these proteins were OM proteins (Fig. 2). BOMP predicted two proteins containing one and two bbtms, where as PSORTb predicted one of them as extracellular protein and the other hypothetical protein was predicted to be from an unknown localization site. Hence these results were compared with TMB-hunt. These two proteins were also predicted by TMB-hunt as integral membrane proteins with scores of 0.62 and 0.73 (see Supplemental. Table 1). From these results, it is clear that these OM proteins were predicted by different bioinformatics programs. Earlier studies established that some membrane protein preparation methods are better over others;34 it is increasingly becoming clear that different methods should be combined to identify the membrane proteome.
Venn diagram depicting the overlapping and uniquely predicted OM proteins using BOMP and PSORTb in P. syringae Lz4W. The commonly predicted proteins which strongly support their presence in OM were shown in the dark portion.
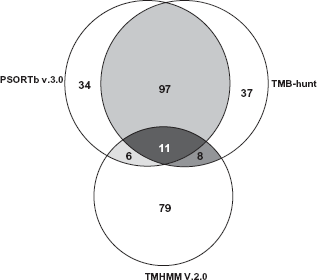
The program of TMB- Hunt predicted 153 proteins containing bbtm. From these 108 proteins were also predicted as OM proteins as revealed by PSORTb (Fig. 3). A major advantage with TMB-Hunt is that it does not rely on β-strand detection. Thus it will be invaluable in complementing other physicochemical and homology based methods. Thus, it also helps in validating the scope of predicting the OM proteins. In addition, both programs BOMP and TMB-Hunt predict the presence of β-barrel transmembrane domains. A comparison of TMB-Hunt and BOMP revealed that 50 proteins were predicted by both the programs. Twelve proteins were uniquely predicted by BOMP, whereas 103 protein were uniquely predicted by TMB-Hunt (Supplemental Table 1). TMB-Hunt cannot discriminate between beta strands of bbtm proteins and some globular proteins. Hence, TMB-Hunt and BOMP are used as additional tools to verify and support PSORTb OM predictions. Thus using these different algorithms it is seen that PSORTb predicted OM proteins efficiently from P. syringae Lz4W.
Venn diagram depicting the overlapping and uniquely predicted membrane proteins of P. syringae Lz4W. Using these methods 148 proteins predicted by PSORTb, 153 proteins by TMB-hunt.108 proteins predicted by PSORTb contain bbtm by TMB-hunt, strongly supporting their location in OM. The CM proteins are uniquely predicted by TMHMM.
Transmembrane helices were also predicted using the hidden Markov model (TMHMM) as described earlier.30 This program can predict the transmembranes helices upto an accuracy of about 98%.35 Comparative results of different programs used for the prediction of α-helices and β-barrels along with the subcellular localization for the membrane proteins of P. syringae were shown in Figure 3. From these results 153 proteins could be identified as membrane proteins containing transmembrane β-barrels and 104 proteins contain helices. In the present study, it is observed that some of the OM proteins predicted by PSORTb generally contain one transmembrane helix, whereas the cytoplasmic membrane proteins contain two and more than two transmembrane domains.
Several heat shock proteins and chaperones were identified from the membrane fraction prepared from P. syringae Lz4W (supplemental Table1). The role of chaperones in cold adaptation has been evidenced earlier.36 Aspartate amino transferase, a protein detected in the membrane fraction in the present study has been shown to play a role in bacterial cryotolerance.37 Several other proteins shown to be involved in cold adaptation of bacteria38 have also been identified in this study.
In conclusion, it appears that preparation of membrane proteins using our protocols is adequate for enriching outer membrane proteins. Using a combination of biochemical methods for the preparation of membrane proteins and bioinformatics algorithms, the membrane proteins of the gram-negative bacteria could be localized. These studies also call for expanding the scope of bioinformatics algorithms for predicting subcellular locations in bacterial cells. Some proteins shown to be playing an important role in cold adaptation were also identified.
Disclosure
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Acknowledgements
MVJ thank Department of Biotechnology (DBT), India for the financial support (BT/PR7383/BRB/10/474/2006) to carry out this research work. We would like to thank Dr MK Chattopadhyay and Dr. Sivakama Sundari for their help in the preparation of the manuscript. Ms Saranya from Bharatidasan University, Thiruchirapalli, carried out this research work at CCMB, Hyderabad in partial fulfilment of the requirements for the award of M. Sc degree in Life sciences.
References
1.
WashburnM.P., WoltersD., YatesJ.R.Large scale analysis of the yeast proteome by multidimensional protein identification technology.Nat Biotechnol.2001; 19: 242–47.
2.
WuC.C., MacCossM.J., HowellK.E., YatesJ.R.A method for the comprehensive proteomic analysis of membrane proteins.Nat Biotechnol.2003; 21: 532–38.
3.
PattersonS.D., AbersoldR.H.Proteomics: the first decade and beyond.Nat Genet.2003; 33: 311–23.
4.
McCormackA.L., SchieltzD.M., GoodeB.Direct analysis and identification of proteins in mixtures by LC/MS/MS and database searching at the low-femtomole level.Anal Chem.1997; 69: 767–76.
5.
JagannadhamM.V.Identification of proteins from membrane preparations by a combination of MALDI TOF/TOF and LC coupled ion trap MS analysis of an Antarctic bacterium Pseudomonas syringae Lz4W, a strain with unsequenced genome.Electrophoresis.2008; 29: 4341–50.
6.
JagannadhamM.V., Abou-EladabE.F., KulkarniH.M.Identification of outer membrane proteins from an Antarctic bacterium.Pseudomonas syringae Lz4W. M110.004549. First Published on March 29, 2011, doi: 10.1074/mcp. M110.004549 (In Press).
7.
MolloyM.P.Two dimensional electrophoresis of membrane proteins using immobilized pH gradients.Anal Biochem.2000; 280: 831–40.
8.
RabilloudT.Membrane proteins and proteomics: love is possible, but so difficult.Electrophoresis.2009; 30: S174–80.
9.
SantoniV., MolloyM.P., RabilloudT.Membrane proteins and proteomics: un amour impossible?Electrophoresis.2000; 21: 1054–70.
FellerG., GerdyC.Psychrophilic enzymes: hot topics in cold adaptation.Nature Mocrobiol Rev.2003; 1: 200–8.
12.
JagannadhamM.V., Jayathirtha RaoV., ShivajiS.The major carotenoid pigment of a Psychrotrophic Micrococcus roseus: purification, structure and interaction of the pigment with synthetic membranes.J Bacteriol.1991; 173: 7911–17.
13.
JagannadhamM V., ChattopadhyayM.K., ShivajiS.The major carotenoid pigment of a psychrotrophic Micrococcus roseuss train: fluorescence properties of the pigment and its binding to membranes.Biochem Biophys Res Commun.1996; 220: 724–28.
14.
JagannadhamM.V., NarayananK., Mohan RaoC., ShivajiS.In vivo characteristics and localization of carotenoid pigments in psychrotrophic and mesophilic Micrococcus roseus using photoaccoustic spectroscopy.Biochem Biophys Res Commun.1996; 227: 221–26.
15.
ChattopadhyayM.K., JagannadhamM.V., VairamaniM., ShivajiS.Carotenoid pigments of an Antarctic bacterium Micrococcus roseus: temperature dependent biosynthesis, structure and interection with synthetic membranes.Biochem Bipohys Res Commun.1997; 239: 85–90.
16.
JagannadhamM.V., ChattopadhyayM.K., SubbalakshmiC.Caratonoids of an Antarctic psychrotolerant bacterium Spingobacteriumantarcticus and a mesophilic bacterium Spingobacteriummultivorum.Arch Microbiol.2000; 173: 418–23.
17.
RayM.K., KumarG.S., ShivajiS.Phosphorylation of membrane proteins in response to temperature in an Antarctic Pseudomonas syringae.Microbiology.1994; 140: 3217–23.
18.
JagatapP., RayM.K.Studies on the cytoplasmic protein tyrosine kinase activity of the Antarctic bacterium Pseudomonas syringae.FEMS Micro Biol Lett.1999; 173: 379–88.
19.
MolloyM.P., HerbertB.R., SladeM.B.Prpteomic analysis of the Escherichia coli outer membrane.Eur J Biochem.2000; 267: 2871–81.
20.
GotohN., WhiteN.J., ChowagalW., WoodsD.E.Isolation and characterization of outer membrane proteins of Burkholderia (Pseudomonas) pseudomonalli.Microbiology.1994; 140: 797–805.
21.
YuN.Y., WagnerJ.R., LiardM.R.PSORTb 3.0; improved protein subcellular localization prediction with refined localization sub categories and predictive capabilities for all prokaryotes.Bioinformatics.2010; 26: 1608–15.
ViklundH., ElofssonA.Best alpha-helical transmembrane protein topology predictions are achieved using hidden MARKOV models and evolutionary information.Protein Sci.2004; 13: 1980–17.
24.
GardyJ.L.PSORTb 2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis.Bioinformatics.2005; 21: 617–23.
25.
BervenF.S., FlikkaK., JensenH.B., EidhammerI.BOMP: a program to predict integral β-barrel outer membrane proteins encoded within genomes of Gram-negative bacteria.Nucleic Acid Res.2004; 32: W394–9.
26.
StruyveM., MoonsM., TommassenJ.Carboxy-terminal phenylalanine is essential for the correct assembly of a bacterial outer membrane protein.J Mol Biol.1991; 218: 141–8.
27.
WimleyC.W.Towards genomic identification of beta-barrel membrane proteins: composition and architecture of known structures.Protein Sci.2002; 11: 301–12.
28.
GarrowA.G., AgnewA., WestheadD.R.TMB-Hunt: an amino acid composition based method to screen proteomes for beta-barrel trans membrane proteins.BMC Bioinformatics.2005; 6: 56.
29.
SonnhammerE.L.L., von HeijneG., KroghA.“A hidden Markov model for predicting transmembrane helices in protein sequences”. In: GlasgowJ, LittlejohnT, MajorFR.LathropR, SankoffDC, SensenCM, editors. Proc of Sixth Int Conf on Intelligent Systems for Molecular Biology. P.CA: AAAI Press; 1998: 175–182.
30.
PautschA., SchulzG.E.High resolution structure of the OmpA membrane domain.J Mol Biol.2000; 298: 273–82.
31.
GaldieroS., GaldieroM., PedoneC.beta-barrel membrane bacterial proteins: structure, function, assembly and interaction with lipids.Current protein pept Sci.2007; 8: 63–82.
32.
HobbR.L., FieldsJ.A., BurnsC.M., ThompsonS.A.Evaluation of procedures for outermembrane isolation from Campylobacter jejuni.Micribiology.2009; 155(Pt 3): 979–88.
33.
TheinM., SouerG., ParamasivamN., GrinI., LinkeD.Efficient sub fractionation of Gram–negative bacteria proteomics studies.J Proteome Res.2010; 9: 6135–47.
34.
KroghA., LarssonB., Von HeijneG., SonnhammerE.L.L.Predicting transmembrane topology with a hidden Markov model: application to complete genomes.J Mol Biol.2001; 305: 567–80.
35.
StrocchiM., FerrerM., TimmisK.N., GolyshinP.N.Low temperature induced systems failure in Escherichia coli: insights from rescue by cold adapted chaperons.Proteomics.2006; 6: 193–206.
36.
SundereswaranV.R., SinghA.K., DubeS., ShivajiS.Aspartate amino transferase is involved in cold adaptation in psychrophillic Pseudomonas syringae.Arch Microbiol.2010; 192: 663–72.
37.
ChattopadhyayM.K.Mechanism of bacterial adaptation to low temperature.J Biosci.2006; 3: 157–65.