
Abstract
The cannabinoids are members of a deceptively simple class of terpenophenolic secondary metabolites isolated from
History of Cannabis Sativa
With the rise of research on natural products and the isolation of alkaloids such as morphine and cocaine, from the opium poppy and coca plant, respectively, cannabis was thought to possess similar chemical constituents. Much of the early research conducted on cannabis and hemp oil cantered on the search for alkaloids and other amine natural products and attempts to develop colorimetric tests for cannabinoids.
2
The search for psychoactive compounds in cannabis, however, led not to a mixture of alkaloids but to the discovery of new terpenes. Most isolation experiments followed a similar procedure for nearly 100 years; hemp oil would be extracted with organic solvents, filtered, followed by removal of the solvent and fractional distillation of the resulting residue.
3
This residue, referred to as
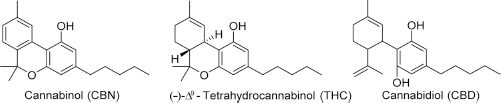
Structures of cannabinol, Δ 9 -THC, and cannabidiol.
It was initially thought that cannabinol was the active constituent of cannabis, but these early reports, likely obtained using impure cannabinol extracts, were proved erroneous in the 1930s.3,8 From 1940 until the 1960s, several other noncannabinoid natural products in cannabis were isolated, including cannabidiol (CBD). 9 The active component of cannabis was finally discovered in 1964 by Gaoni and Mechoulam, with the report of the structure and partial synthesis of (–)-Δ 9 -tetrahydrocannabinol (THC). 10 The discovery of new compounds in cannabis has continued, with over 100 phytocannabinoids reported to date.
Phytocannabinoids
Phytocannabinoids are found throughout all major morphologies of cannabis. Cannabinoids are mixed polyketides derived from malonyl-CoA, hexanoyl-CoA units prenylated with geranyl phosphate.11–13 This biosynthetic pathway, shown in Figure 2 with the synthesis of CBD, THC, and CBN, produces several classes of phytocannabinoids.
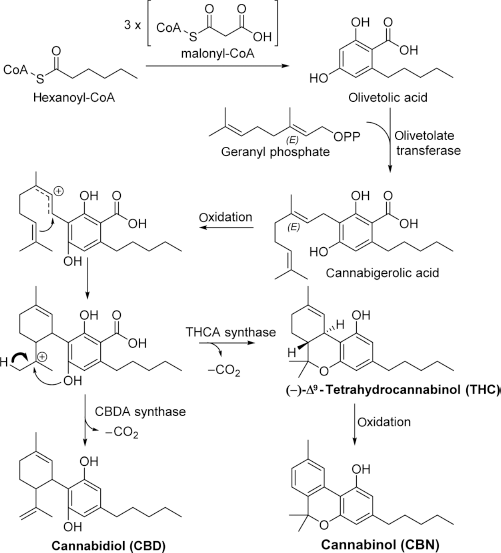
Phytocannabinoid biosynthesis.
Of the more than hundred phytocannabinoids isolated and characterized, THC and CBD, depicted in Figure 1, have received the most attention in both basic science and clinical research. THC is marketed as dronabinol (Marinol®) and is currently approved for the treatment of anorexia in Acquired Immune Deficiency Syndrome (AIDS) patients and chemotherapy-induced nausea and vomiting. 14 CBD has not been approved by the US Food and Drug Administration (FDA), but clinical trials are underway exploring the use of CBD, branded as Epidiolex®, in the treatment of epilepsy and Dravet syndrome, a severe seizure disorder in children.15,16 Despite the long-standing traditional medicinal uses of cannabis, and the culmination of scientific evidence leading to FDA approval of THC, the mechanism of action of cannabinoids in humans remained a conundrum until recently. The cannabinoid (CB) receptors remained elusive for 30 years after the discovery of THC. Both CBD and THC exert their therapeutic effects through the CB receptors, in addition to G-protein-coupled receptor 55 (GPR55), 5-hydroxytryptamine (5-HT)-3A ligand-gated ion channel, transient receptor potential cation channel Ankyrin type 1 (TRPA1), and transient receptor potential cation channel vanilloid type 1 (TRPV2).17–20 The pharmacology of CBD is not entirely understood with respect to the treatment of seizures; CBD has been shown to block both CB receptors, activate several TRP cation channels, and activate the 5-HT1A receptor.18,19,21
CB Receptors and the Endocannabinoid System
The G-protein-coupled receptor (GPCR) superfamily of genes encodes for 800 GPCRs. This superfamily of genes is further divided into five major families: glutamate, rhodopsin, adhesion, frizzled/taste2, and secretin. 22 The largest of these is the rhodopsin family, containing 672 GPCR genes, approximately 300 of which encode known GPCRs, with the remainder classified as orphan receptors whose structure, endogenous ligand(s), and function remain unknown.23–25 GPCRs are characterized by having a transmembrane domain unit with seven alpha-helices coupled to a G-protein consisting of three subunit proteins: Gα, β, and γ. Upon binding of an agonist ligand to the transmembrane domain, the G-protein subunits catalyze downstream functions by coupling to another cellular protein (eg, adenylyl cyclase, protein kinases, etc.). 26 Because of their significant role in human cellular functions, drugs targeting GPCRs make up 30%–40% of all drugs on the market. 27
The CB receptors are members of the rhodopsin-like family of GPCRs. The first evidence of a CB receptor surfaced in 1984, with Howlett et al demonstrating that select cannabinoids decreased cyclic adenosine monophosphate (cAMP) concentrations in neuroblastoma cells.28,29 Further work in 1986 showed that cannabinoids induce a decrease in cAMP production, and this effect was eliminated by exposing cells to pertussis toxin, a known Gαi (commonly referred to simply as the Gi protein) protein inhibitor, which strongly suggested the presence of a CB binding GPCR. 30 In 1988, the same group characterized a CB-specific receptor in rat brain, and in 1990, the CB1 receptor was finally cloned from a cDNA library from rat cerebral cortex tissue. 31 In the same year that CB1 was discovered, tissue distribution studies showed that CB1 was one of the most abundantly expressed receptors in the brain, nearly equivalent to the expression of glutamate and GABA receptors.32,33 The correlation between CB1 localization in the brain and the known pharmacological effects of CB agonists was made immediately clear; CB1 expression in the basal ganglia and cerebellum was associated with the effects on gait, and expression in the cerebral cortex and hippocampus was associated with the effects on cognition and memory. More recent studies have found CB1 expression in the spleen, tonsils, gastrointestinal tract, uterus, prostate, vascular smooth muscle cells, and adrenal glands. 34
While the CB1 receptor was commonly referred to as the
These and many other results, however, have been called into question, as anti-CB2 antibodies used in these immunohistochemical methods have been demonstrated to have nonspecific binding with other proteins.42,43 The immunomodulatory role of CB2 has remained unchallenged, and CB2 has been heavily implicated in neurodegenerative diseases such as Huntington's and Alzheimer's diseases.44,45 Increased expression of CB2 in the brain was confirmed with CB2-selective positron emission tomography (PET) tracers in Alzheimer's mice models; this increased expression was concomitant with the formation of amyloid-beta plaques, suggesting a potential utility for CB2 PET tracers as diagnostic for the onset of neuroinflammation.
Activation of either CB1 or CB2 produces a dose-dependent decrease in cellular cAMP levels and modulation of intracellular Ca2+ and K+ levels. 46 Stimulation of CB receptors results in activation of the p42/44 mitogen-activated protein kinases (MAPK), otherwise known as the extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2), respectively, as well as p38 MAPK and c-Jun N-terminal kinases.47,48 Signal transduction studies have linked this CB1/2 mediation of ERK1/2 to downstream regulation of genes, controlling cytokine synthesis, transcription regulation, and cell differentiation (Fig. 3).49,50
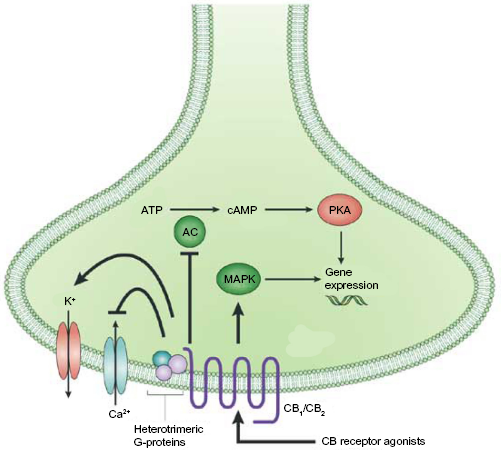
Neuronal CB signaling. Activation of a CB receptor with an agonist causes several downstream effects: inhibition of adenylcyclase and inwardly rectifying calcium channels, and activation of potassium channels as well as the mitogen-activated protein kinase pathway. Activation of MAPK modulates gene expression, depending on downstream signaling, cell types, etc. Gene expression can also be modulate as a downstream effect of adenylyl cyclase inhibition through the activation of protein kinase A.
Endocannabinoid System
While the discovery (and the naming) of the CB receptors was driven by a desire to understand the pharmacological effects of cannabis, both receptors are involved in extensive signaling pathways known as the endocannabinoid system. The presence of CB GPCRs suggested the existence of endogenous ligands, and as most phytocannabinoids are highly lipophilic, it was assumed that these ligands would likely be lipids.
The identification of anandamide (AEA) by the Mechoulam group in 1992 confirmed its role as an endogenous ligand for the CB receptors, with a
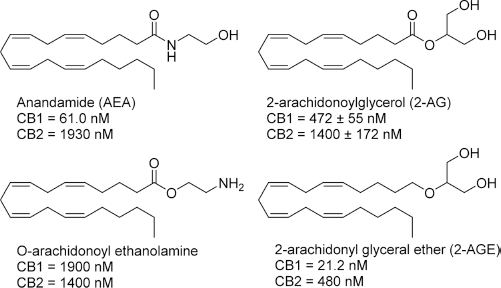
Structures and CB receptor affinities of major endocannabinoids.
For a very broad overview, endocannabinoid signaling typically occurs in retrograde fashion, from post- to presynaptic neurons (Fig. 5). Due to the highly hydrophobic nature of endocannabinoids like 2-AG, it was initially thought that endocannabinoids were synthesized in the same cell in which receptor binding occurs. This was supported by the observation that endocannabinoids could approach a receptor by moving laterally through the cell membrane, through postsynaptic nonretrograde signaling. 64 However, the identification of AEA in interstitial fluid and cell incubation media suggested additionally that AEA, and likely other endocannabinoids, can travel across a synapse by either passive diffusion or active transport mechanisms, although a specific mechanism has yet to be resolved.65–68
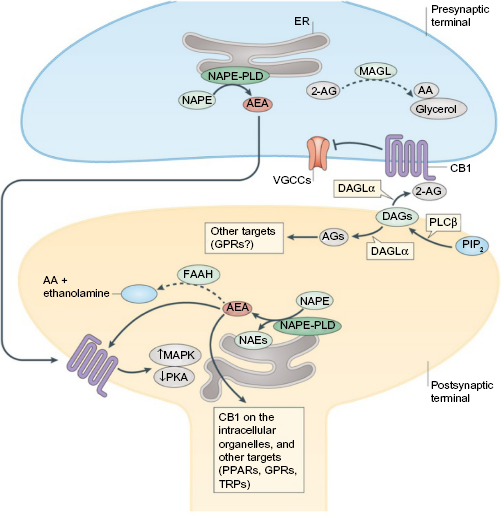
Synaptic endocannabinoid signaling. Dashed arrows indicate inactivation of endocannabinoid.
In retrograde signaling, endocannabinoids cause a variety of downstream effects. Presynaptic CB1 activation causes two major neurotransmitter inhibition mechanisms: short-term and long-term plasticity. 69 In short-term plasticity, elevation of the intracellular Ca2+ levels by postsynaptic depolarization stimulates the production of 2-AG, which diffuses across the neuronal synapse to CB1 receptors on the presynaptic neuron.70–72 Activation of CB1 causes inhibition of Ca2+ influx via voltage-gated Ca2+ channels and subsequent downregulation of neurotransmitter release. Long-term plasticity, while initiating in a similar fashion, involves the suppression of neurotransmitter release via the downregulation of cAMP production and protein kinase A inhibition.
Once released, endocannabinoids are rapidly deactivated by two enzymes: fatty acid amide hydrolase 1 and monoacylglycerol lipase. 44 The distribution of these enzymes provides some evidence as to the signaling mechanisms carried out by the endocannabinoid system, as fatty acid amide hydrolase 1 is located postsynaptically and monoacylglycerol lipase presynaptically.
The role of CB2 is less well defined than CB1 in the endocannabinoid system. The involvement of CB2 in the endocannabinoid signaling system has been relegated to that of an immunomodulatory mediator. Like CB1, CB2 also decreases the production of cAMP, although to a lesser degree, and unlike CB1, it has not been found to be coupled to G proteins other than Gi, somewhat limiting its inhibitory effect on Ca2+ and K+ channels. 36 Also unique to the activation of CB2 receptors is an initial decrease in cAMP production, followed by a sustained increase up to 10-fold in T-cell cAMP levels, which can lead to suppression of T-cell signaling, manifesting phenotypically as an immunosuppressant effect. 73 Immuno-histochemical and mRNA analyses show CB2 localization to occur primarily in microglial cells in the brain, neutrophils, macrophages, monocytes, and lymphocytes peripherally, with significantly increased expression under inflammatory conditions.41,74,75 Increased endocannabinoid production in immune cells has been linked to pro-inflammatory stimuli and hematopoietic stem cell differentiation.76,77 For a more in-depth discussion of endocannabinoid signaling, several timely reviews have been published.68,78–82
Synthetic Classical Cannabinoids
Since the initial discovery of THC and other related cannabinoids, numerous modifications and analogs were synthesized in an attempt to define the structure–activity relationship (SAR) of THC at both CB1 and CB2. Δ9-THC contains five major structural features, depicted in Figure 6: the C3 side chain, phenolic hydroxyl, and three rings: the aromatic A-ring, pyran B-ring, and cyclohexenyl C-ring. While not present in any natural cannabinoids, some important synthetic analogs replace the pyran B-ring with a substituted aliphatic chain, and as such, the
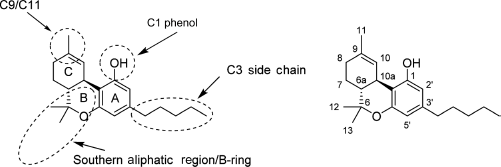
SAR of classical cannabinoids (left) major pharmacophores of classical cannabinoids, and common regions of functionalization and analog synthesis; (right) dibenzopyran numbering of Δ9-THC.
Many of the earliest SAR studies on classical cannabinoids involved modifications to the C3 side chain. While most analogs contain saturated straight or branched alkyl chains, a number of C3 side chains incorporating unsaturated alkyl chains, heteroatoms, and functional groups such as esters, carboxylic acids, ethers, nitriles, and heterocycles were reported, with varying effects on CB1 and CB2 potency and selectivity.
C3 alkyl analogs
The length of the C3 side chain of THC directly correlates with CB1 and CB2 binding affinity; an increase in chain length leads to an increase in binding affinity at both receptors (Fig. 7). CB binding affinity data for methyl- or ethyl-substituted THC analogs have not been published; however, a study conducted in 2011 examined the functional activity of these THC analogs, demonstrating decrease in the receptor affinity in a linear fashion with decreasing chain length. 83 Interestingly, this study found an inverse relationship between chain length and TRPA1 channel activity; a one-carbon chain was found to be a potent TRPA1 agonist, this effect decreasing with additional carbons on the chain. This may be a contributing factor to the biological significance of cannabinoids like propyl-substituted Δ9-tetrahydrocannabivarin, which does not activate either CB receptor yet retains numerous biological effects.
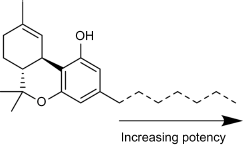
Relationship between alkyl chain length and CB receptor binding affinity.
Utilizing the C3 alkyl chain as a point of diversification in classical cannabinoids has remained a staple of CB research since the discovery of Δ9-THC. Numerous analogs, containing a variety of carbon chains and rings with and without heteroatom incorporation, provided a well-defined and predictable SAR profile for this portion of the THC scaffold.
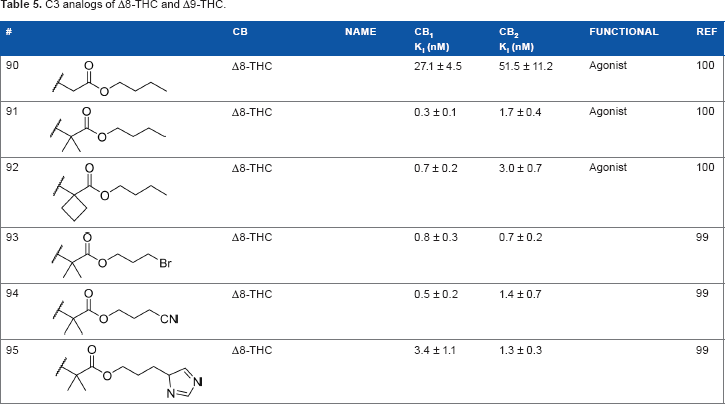
Of the major pharmacophores defined for classical CB SAR, the C-3 side chain seems to have the largest influence on binding affinity for the CB receptors. Table 1 illustrates a homolog series of Δ8-THC and Δ9-THC analogs with various saturated aliphatic substitutions. Entries
C3 alkyl analogs of Δ8-THC and Δ9-THC (human CB1 and CB2).
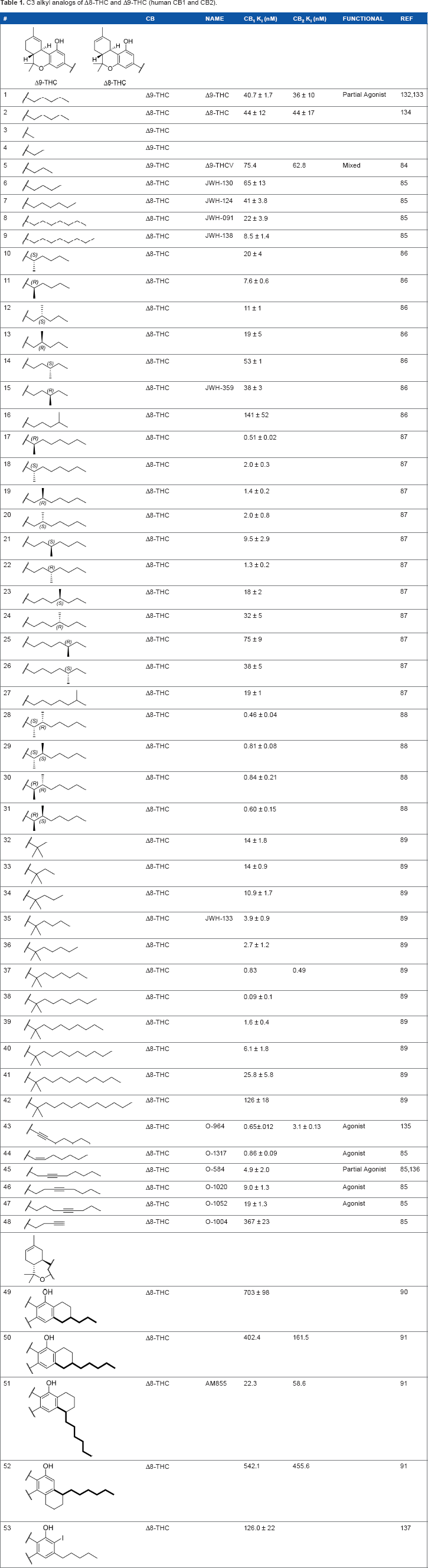
Introducing unsaturation on the alkyl chain does not significantly modulate CB1 receptor binding, with activity retained between homologs of the same chain length [eg, compare the unsaturated 8-carbon analogs
A series of rotationally restricted Δ
8
-THC analogs, entries
These aliphatic side chain analogs offer several conclusions that can be inferred with regard to CB receptor binding requirements. A 5–8 carbon length alkyl chain is optimal, with binding affinity decreasing when shorter and longer alkyl chains are incorporated. Restricting the flexibility of the alkyl chain lends some insight into the optimal conformation for receptor binding. First, introducing methyl substitutions in various positions on the chain adds steric congestion that restricts the number of possible rotamers, in addition to providing hydrophobic bulk that may be interacting with the receptors. Branching close to the aromatic ring provides a minimum 10-fold increase in affinity for CB1 (
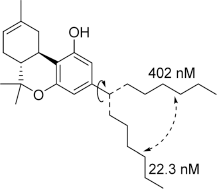
Rotational conformers of 3-heptyl-Δ8-THC. Studies of rotationally restricted alkyl chains determined that the optimal conformation of the chain is oriented downward away from the phenol, rather than linearly away from the phenyl ring.
To investigate the SAR requirements of C1' substitutions on the side chain, several series of compounds containing aliphatic rings and heterocycles were synthesized, as illustrated in Table 2. Transforming the 1',1'-geminal dimethyl substitution into a compact cyclopropyl ring further enhances activity at both receptors. 94 Expanding the ring size to 4, 5, and 6 carbons modulates the activity as a function of the lowest energy conformation of the alkyl chain, as well as the presence of hydrophobic bulk, potentially creating steric clashes with the putative binding site in the receptor. Quantitative structure–activity relationship studies confirm that the cyclopropyl and cyclopentyl rings force the chain into similar favorable orientations, with the cyclobutyl- and cyclohexyl-substituted chains adopting less favorable conformations for optimal receptor binding. 95 This orientation, with the alkyl chain oriented perpendicular to the aromatic ring of the THC scaffold, appears to be the optimal conformer for receptor binding.
C3 analogs of Δ8-THC and Δ9-THC.
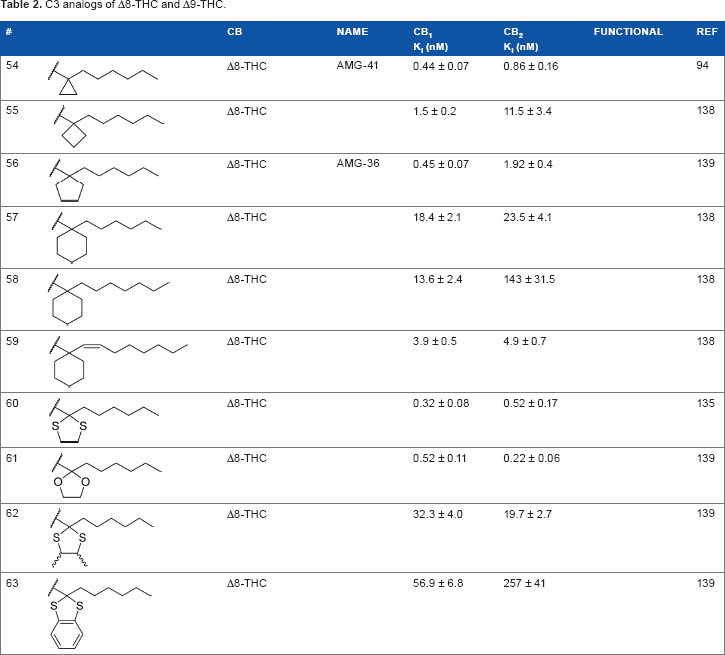
In addition to the alkyl chain conformation, the increased affinity of these compounds also suggests the presence of a hydrophobic binding subsite near the phenyl ring in both CB1 and CB2. Both receptors appear to be indifferent to heteroatoms at this position, illustrated by the high affinity of analogs
The replacement of the alkyl chain with various ring structures results in a variety of effects on receptor binding (Table 3). To explore the necessity of hydrophobic bulk near the phenolic ring, a series of saturated alkyl ring analogs were synthesized and evaluated. Bulky bornyl and adamantyl derivatives
C3 analogs of Δ8-THC and Δ9-THC.
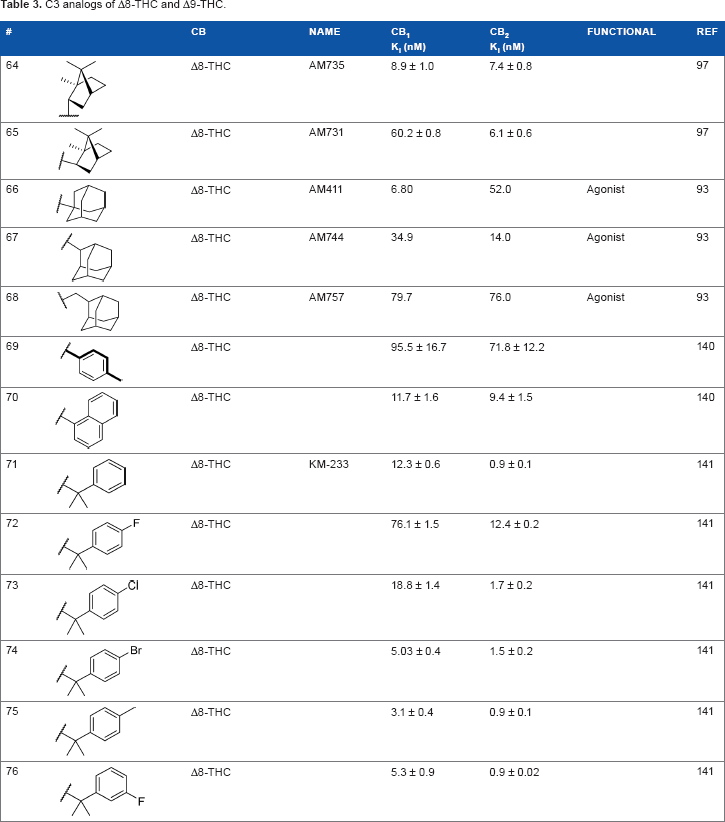
Using more planar phenyl rings, analogs
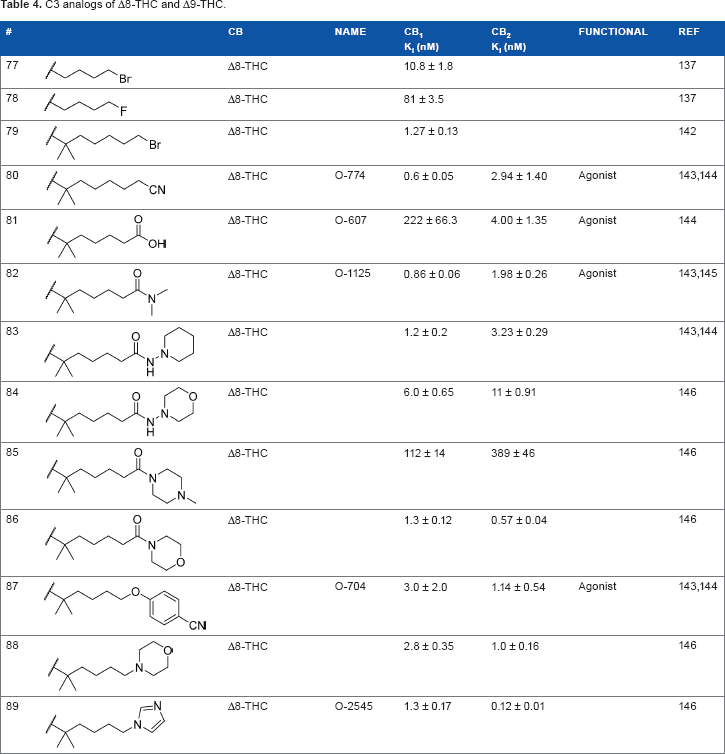
The addition of heteroatom substitutions on the terminal position of the alkyl chain provides analogs that display increases in binding affinity as well as enhanced polarity (Table 4). The use of halogens and nitrile pseudohalogens on the terminal position of the alkyl chain for both the pentyl (
C3 analogs of Δ8-THC and Δ9-THC.
In another effort to improve the pharmacokinetics and bioavailability of classical type cannabinoids, Nikas et al.
99
and Sharma et al.
100
synthesized a series of side chain analogs containing a labile ester, in order to increase polarity and solubility and to extend the half-life
C3 analogs of Δ8-THC and Δ9-THC.
C1 phenol analogs
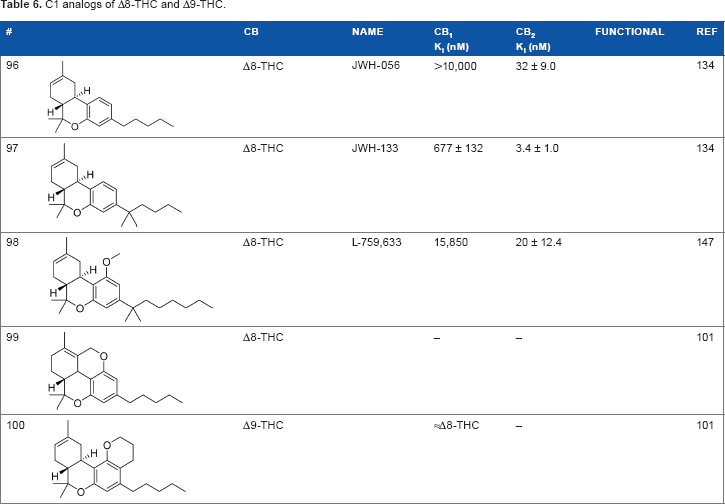
Another major point of structure modification on the THC scaffold is the C1 phenol (Table 6). Analogs lacking the phenolic hydroxy group or those that have been modified with minor changes to the phenolic group can result in drastic changes in the pharmacological activity of these compounds. It was quickly realized that etherification or removal of the phenol generated compounds that displayed significant selectivity for CB2. For example, the deoxy-Δ8-THC analog
C1 analogs of Δ8-THC and Δ9-THC.
Analogs
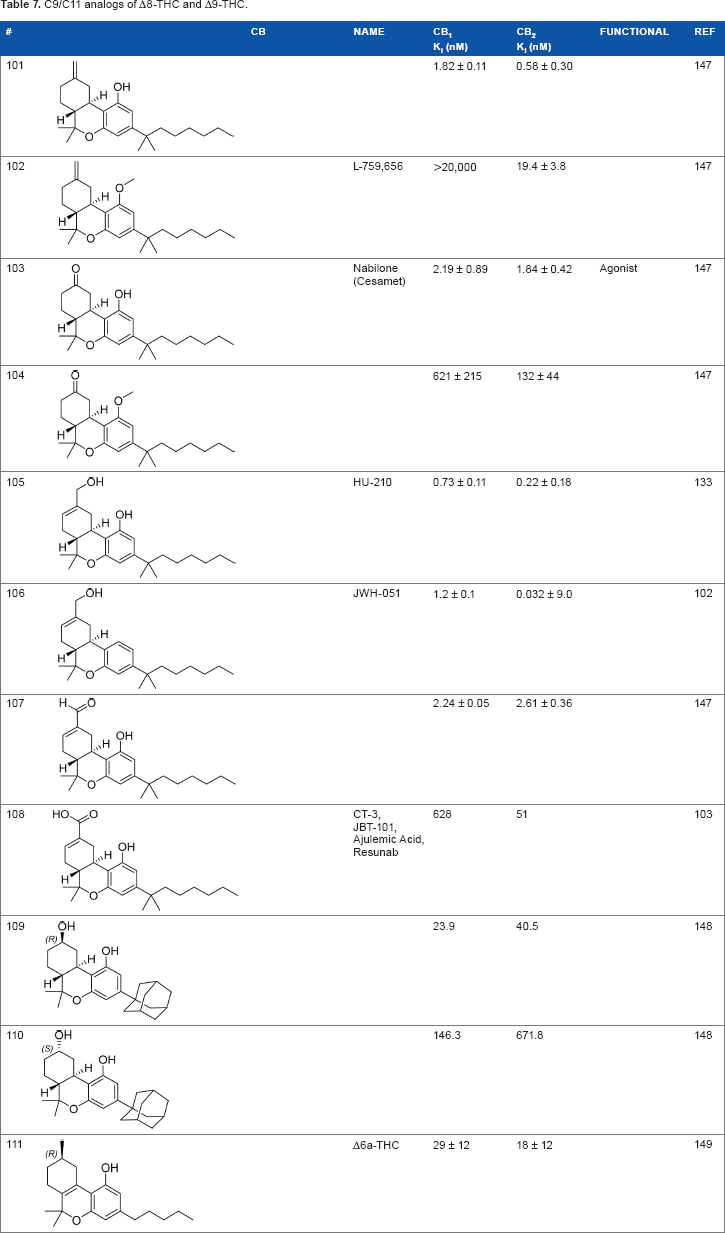
C9/C11 analogs
The C11 methyl group is another major pharmacophore at which minor structural changes can significantly modulate receptor binding (Table 7). Substitutions at this position do not confer selectivity when compared to analogs modified at the C1 phenol; however, binding affinity can be greatly enhanced. Methylene analogs
C9/C11 analogs of Δ8-THC and Δ9-THC.
Miscellaneous analogs
Early SAR studies revealed that the pyran ring was not a requirement for cannabinergic activity in animal assays. Subsequent work on the core structure of
Miscellaneous analogs of Δ8-THC and Δ9-THC.

Nonclassical Cannabinoids
CB1 selective
While classical CB development has remained a staple of compound synthesis, compounds adopting alternative scaffolds have become prominent. The lipophilicity of most classical cannabinoids has hampered their development into viable drugs, so efforts to increase the polarity and water solubility of new CB ligands has been an area of major focus.
Pravadoline (WIN 48,098) was a cyclooxygenase (COX) inhibitor developed by Bell et al. at Sterling-Winthrop in the 1980s, but its antinociceptive activity was significantly higher than other known COX inhibitors, despite its lower effect on prostaglandin synthesis compared to known nonsteroidal anti-inflammatory drugs. 107 With animal assay results similar to known cannabinoids, the possibility was raised that the antinociceptive activity was derived not from COX binding, but from CB receptor activity. Structural optimization of pravadoline yielded one of the first nonclassical cannabinoids, WIN 55,212-2. 108 Like CP-55,940, WIN 55,212-2 is a CB receptor agonist widely used in CB receptor assays as a reference compound. Since the discovery of WIN 55,212-2, numerous other potent, nonclassical cannabinoids were synthesized; two other notable compounds in this class are depicted in Figure 9. The (aminoalkyl)indole JWH-018 is nearly equipotent at both CB receptors, with slight selective toward CB2. 109 ' 110 In recent years, JWH-018 has gained notoriety for its illicit use in herbal blends known as “Spice.”111,112 Diarylpyrazole derivative SR141716 was first described as a CB1 receptor antagonist, but further studies found it to be a CB1 inverse agonist. SR141716, also known as rimonabant, was briefly approved and marketed as Acomplia by Sanofi in Europe as an antiobesity treatment from July 2006 to October 2008, when significant side effects such as suicidal thoughts and depression forced its withdrawal from the market. 113
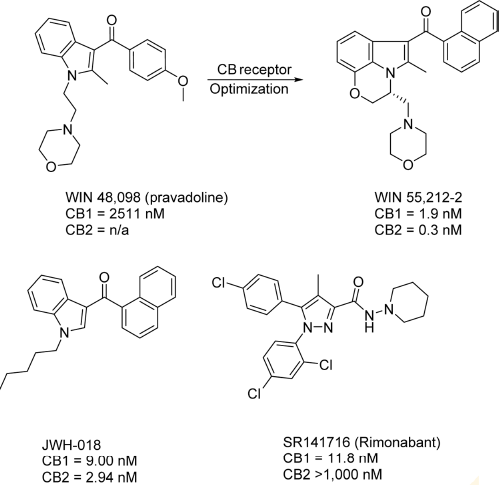
Representative nonclassical CB1 ligands. The first nonclassical CB scaffold discoveries were the
CB2 selective
CB2-selective agonists have been explored for a number of therapeutic indications, most commonly as analgesic and anti-inflammatory compounds (Fig. 10). Cannabinor, formerly known as PRS-211,375, was tested in humans for the treatment of several modes of pain; it ultimately failed due to a lack of efficacy in a Phase IIb study in the treatment of pain following third molar tooth extraction. 114 Cannabinor may have also suffered from modulation of bladder activity, as animal studies have demonstrated a CB receptor-linked effect increasing urination frequency.115,116 GW-842,166X was also explored for the treatment of third molar tooth extraction and osteoarthritis, but failed in Phase II trials due to lack of efficacy.117,118 S-777,469 completed Phase II trials for atopic dermatitis in 2011, but no clinical data or future plans for clinical testing of this compound have been released, indicating that its development may have been halted.119–121
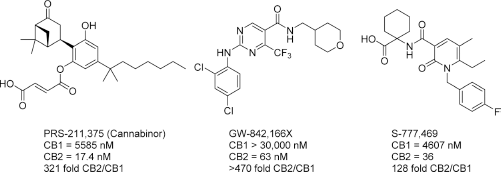
CB2-selective agonists. PRS-211,375 and GW-842,166X were both investigated in clinical trials for the treatment of pain; both failed for lack of efficacy and presence of adverse side effects. S-777,469 completed Phase II trials, but no results have been reported and development has presumably halted.
After several failed clinical trials, the possibility that years of immunostaining results were inaccurate and halted CB2 research programs, the progress of CB2-targeted therapeutics was seemingly at an impasse. Research efforts into therapeutic potential of CB2 selective agonists have recently focused on improving selectivity by several orders of magnitude. Although many of the conventional CB2 agonists were as much as 500-fold selective, this may not be effective
Since late 2014, several landmark improvements have been reported in the pharmacological tools used to study the CB2 receptor. Transgenic mice expressing a CB2-GFP reporter have allowed for highly specific and accurate studies of CB2 expression, especially in light of the specificity problems with most anti-CB2 antibodies. 125 Many advances have been made in the development of CB2-selective compounds. New scaffolds have been employed in the synthesis of compounds that display selectivity as high as 35,000-fold, significantly greater than previously reported compounds (Fig. 11).
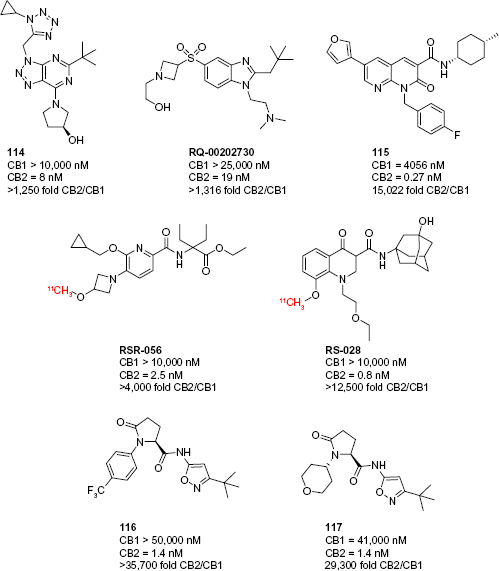
CB2-selective ligands reported in 2014 and 2015.
2015 has been a landmark year for drug discovery programs focusing on the development of selective CB ligands. Triazolopyrimidine
Outlook
Drug discovery programs focusing on CB receptor-targeted therapeutics will continue expanding with the identification and characterization of novel CB receptor-selective ligands. The development of receptor-selective modulators coupled to an understanding of their functional pharmacological activity (agonist; inverse agonist/antagonist) and mode of binding (orthosteric, allosteric) will prove critical for moving candidates to preclinical disease model studies. Classical cannabinoids, including both natural product and semisynthetic derivatives that have been described in this review, have ushered the development of nonclassical synthetic cannabinoids comprising novel scaffolds exhibiting receptor-selective profiles. Compounds selectively modulating the CB2 receptor may have utility in the treatment of inflammation, diabetes, cancer, pain, and other diseases. However, CB2-selective drug candidates thus far have failed in clinical trials due to a lack of efficacy and/or their propensity to mediate CB1 effects, even while displaying several hundred-fold selectivity profiles for CB2 over CB1.
Author Contributions
Wrote the first draft of the manuscript: EWB. Contributed to the writing of the manuscript: EWB, JMR. Agreed with manuscript results and conclusions: EWB, JMR. Jointly developed the structure and arguments for the paper: EWB, JMR. Made critical revisions and approved the final version: EWB, JMR. Both the authors reviewed and approved the final manuscript.