
Abstract
Age-related macular degeneration (AMD) is a major cause of irreversible blindness affecting elderly people in the world. AMD is a complex multifactorial disease associated with demographic, genetics, and environmental risk factors. It is well established that oxidative stress, inflammation, and apoptosis play critical roles in the pathogenesis of AMD. The mitogen-activated protein kinase (MAPK) signaling pathways are activated by diverse extracellular stimuli, including growth factors, mitogens, hormones, cytokines, and different cellular stressors such as oxidative stress. They regulate cell proliferation, differentiation, survival, and apoptosis. This review addresses the novel findings from human and animal studies on the relationship of MAPK signaling with AMD. The use of specific MAPK inhibitors may represent a potential therapeutic target for the treatment of this debilitating eye disease.
Introduction
Age-related macular degeneration (AMD) is the leading cause of blindness in people 60 years and older in developed countries. 1 A recent systematic review and meta-analysis has shown that 8.7% of the worldwide population has AMD, and with the increase of lifespan the projected number of people with the disease will be at about 196 million in 2020, reaching 288 million in 2040. 2 About 1.75 million Americans are affected by AMD, and this number is expected to grow to almost 3 million by 2020. 3 AMD is a complex multifactorial disease that occurs over time and is characterized by degeneration of the retinal photoreceptors, retinal pigment epithelium (RPE), and choroidal neovascularization (CNV). AMD can be classified into two broad groups: dry (nonvascular or atrophic) affects 80%–90% of AMD patients and wet (neovascular or exudative) affects 10%–15%, but is responsible for approximately 90% of AMD-related vision loss. 4 There is no cure, but AMD treatments may prevent severe vision loss or slow the progression of the disease considerably. Several treatment options are available, including anti-VEGF therapy, laser surgery, photodynamic therapy, vitamins, and nutritional supplements.5–8 The etiology of AMD is complex and includes both genetic and nongenetic factors. Genome-wide association studies (GWAS) have revealed common genetic variants at a number of loci. Alterations in genes of the complement system and inflammatory pathways, as well as variations in genes related to oxidative stress, have been associated with AMD.9–12 Among the nongenetic factors, aging, smoking, hypertension, and diet significantly contribute to an increase in the AMD risk. 13 There is ample evidence that oxidative stress is involved in AMD pathogenesis and progression.14,15 Oxidative stress and reactive oxygen species (ROS) have been implicated in the activation of various signaling pathways, including the mitogen-activated protein kinases (MAPKs).16,17 MAPKs are important mediators of signal transduction and play a key role in the regulation of many cellular processes, such as cell growth and proliferation, differentiation, and apoptosis. 18 Several studies suggest that MAPKs are involved in oxidative stress-induced RPE degeneration,19–22 which is described in more detail in the “AMD and MAPK signaling” section. Findings have revealed activation of the MAPK signaling pathways extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK in model systems, including human RPE cell cultures and murine models of AMD.23–28 In addition, linkage disequilibrium-independent genomic-enrichment analysis demonstrated association of AMD with genes encoding the MAPK signaling pathway (JNK, p38, ERK1/2, and ERK5). 29 Moreover, a new software “AMD Medicine” was used to compare the transcriptomes of normal human RPE-choroid and AMD-affected RPE-choroid samples. 30 The results demonstrated activation of several signaling pathways, including ERK in RPE-choroid AMD phenotypes. These data clearly suggest that MAPK signaling is involved in AMD pathogenesis, and MAPK inhibitors could provide a novel therapeutic strategy for prevention or treatment of AMD.
AMD: Pathology, Genetics, and Current Treatments
AMD is the leading cause of blindness in the elderly that damages the central region of the retina (macula). Early stage of the disease is characterized by the presence of medium-sized drusen, which are extracellular deposits containing proteins, lipids, and inflammatory mediators. 31 As the disease progresses, it can develop into intermediate and late AMD. There are two types of late AMD: geographic atrophy and neovascular, or wet AMD. Early and intermediate, as well as geographic atrophy, are generally referred to as dry AMD. Dry AMD is characterized by RPE senescence and geographic RPE loss, while wet AMD is characterized by degeneration of RPE and abnormal growth of pathologic choroidal vessels. Dry AMD is a chronic disease that usually causes some degree of visual impairment, whereas wet AMD could rapidly progress to blindness if left untreated.
AMD is a complex, multifactorial disease of aging. In the aging retina, oxidative stress and ROS have been widely acknowledged to play a major role in the pathophysiology of disease.14,32 The retina is highly susceptible to oxidative stress because of its high consumption of oxygen, high metabolic activity, and exposure to light. Excessive ROS levels can damage lipids, proteins, and nucleic acids. This process subsequently leads to cell death unless it is neutralized by the oxidant defense system. Retinas of patients with AMD show increased oxidative damage and drusen contain high amounts of oxidized proteins, as well as increased content of redox-sensitive proteins.33–35 A growing body of evidence indicates that inflammation plays an important role in both dry and wet AMD.11,36,37 This includes not only mild infiltration of macrophages and accumulation of microglia but also the presence of inflammatory components such as the complement pathway, cytokines, and chemokines. A longitudinal population study provides further support for the role of oxidative stress and inflammation in the pathogenesis of AMD. 38 Angiogenesis, the process of forming new blood vessels, is a hallmark in the pathology of wet AMD. Among the angiogenic factors investigated, the vascular endothelial growth factor (VEGF) has been shown to be a key factor in animal models and AMD patients. Animal models have provided evidence for a relationship between VEGF expression and the development of CNV.39,40 Increased expression of VEGF was found in surgically excited CNV membranes41,42 and increased levels of VEGF were detected in vitreous samples from AMD patients. 43 Although AMD risk involves many factors, it is well recognized that there is a strong genetic contribution, as evidenced by recent GWAS studies. This includes genes associated with the complementary pathway (CFH, CFI, C2/CFB, C3), lipoprotein metabolism (APOE, CETP, LIPC), angiogenesis (VEGFA, TGFBR1), extracellular matrix (TIMP3, COL8A1/FILIP1L, COL10A1), cell death (IER3/DDR1, TNFRSF10A), glucose and lactate transport (SLC16A8, B3GALTL), DNA repair (RAD51B), cleavage of proteoglycans and inhibition of angiogenesis (ADAMTS9/MIR548A2), and mitochondrial gene ARMS2, whose function remains unknown. 12 In addition to the 19 candidate genes based on GWAS, polymorphism in genes encoding chemokines and cytokines have been associated with the risk of AMD development and progression. For greater detail, the reader is referred to the excellent and comprehensive reviews on the genetic component of the disease.9–12
There is no cure for AMD, but several treatment options are available that can prevent severe vision loss or slow the progression of the disease considerably. Currently, three VEGF antagonists such as ranibizumab (Lucentis), bevacizumab (Avastin), and aflibercept (Eylea) are used as the standard treatment for wet AMD. 44 All agents are injected intravitreously and require proper treatment schedule. Another VEGF antagonist, pegaptanib (Macugen) was the first drug approved for wet AMD by the United States Food and Drug Administration in 2004. However, pegaptanib is hardly used anymore because of its limited efficacy compared to the other three drugs, and it is no longer available in some countries. Although the VEGF antagonists are the current standard therapy for wet AMD, they have limitations due to the costs of these drugs, their need for frequent injections, and possible systemic adverse events and ocular complications with repeated high dosages of anti-VEGF compounds. 45 Other treatments for wet AMD include photodynamic therapy, which is less common than anti-VEGF injections and is used mostly in combination with them for specific forms of wet AMD.5,6,44 Thermal laser photocoagulation and macular surgery are less common than other treatments.5,6 In addition, many potential therapeutic strategies focusing on inhibition of the complement pathway are in preclinical trials. 46 In contrast to wet AMD, there is no approved therapy for dry AMD, although a few are now in clinical trials. Current clinical trials are investigating multiple modalities, including drugs that decrease oxidative stress, treatments targeting complement pathway and inflammation, the visual cycle, neuroprotection, and cell replacement therapy. 47 Dry AMD management consists of lifestyle modifications such as quitting smoking and healthy diet supplemented by zinc and antioxidant vitamins (vitamin C, vitamin E, and beta-carotene).
MAPK Signaling
MAPKs are a family of evolutionary well-conserved protein kinases that are expressed in all eukaryotic cells. Members of the MAPK family play a critical role in many cellular processes, including proliferation, differentiation, apoptosis, and survival, among others. In mammals, the MAPK signaling pathways fall into four distinct groups: ERK1/2, JNK1/2/3, p38 (α, β, γ, and δ), and ERK5. 48 They are activated by diverse extracellular stimuli such as growth factors, cytokines, mitogens, hormones, and various cellular stresses including oxidative stress, heat shock, ultraviolet (UV) irradiation, hypoxia, ischemia, and DNA-damaging agents via receptor-dependent and -independent mechanisms. 49 Each group of MAPKs contains a three-tiered kinase signaling cascade: MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK), and MAPK (Fig. 1). MAPKKKs are Ser/Thr protein kinases that are activated through phosphorylation, which, in turn, leads to phosphorylation and activation of MAPKKs, which then stimulate MAPK activity through dual phosphorylation on Thr and Tyr residues within a conserved Thr-X-Tyr motif located in the activation loop of the kinase domain. 50 For example, ERK1/2 and ERK5 have the dual phosphorylation motif Thr-Glu-Tyr, JNK has Thr-Pro-Tyr, and the Thr-Gly-Tyr motif is present in p38 MAPK. 51 Once activated, MAPKs phosphorylate and activate an array of transcription factors present in the cytoplasm and nucleus, leading to the expression of target genes and resulting in a biological response.

Simplified diagram depicting MAPK signaling. In mammals, the four major groups of MAPKs, ERK, JNK, p38, and ERK5 are activated by various extracellular stimuli. Once activated, MAPKs phosphorylate and activate an array of transcription factors.
The ERK pathway is the first MAPK cascade elucidated and the best characterized. 52 ERK1/2 are stimulated in mammalian cells via tyrosine kinase receptors and G-protein-coupled receptors through both Ras-dependent and Ras-independent pathways. 53 They are also activated by growth factors, mitogens, cytokines, osmotic stress, and in response to insulin. Activated Ras-GTP phosphorylate Raf (isoforms A, B, and C), which in turn phosphorylates and activates MEK1/2 and ERK1/2. ERK pathway plays a central role mainly in the control of cell proliferation and differentiation, and also in neuronal plasticity, survival, and apoptosis. 54
The first JNK family member, referred to as p54 MAP-2 kinase, was purified from cycloheximide-treated rat liver, 55 and subsequently cloned and named JNKs because of their ability to phosphorylate and activate the transcription factor c-Jun. 56 There are currently three mammalian JNKs with several isoforms, namely, JNK1, JNK2, and JNK3. 57 It has been shown that JNKs are activated in response to various cellular stresses such as heat shock, ionizing radiation, oxidative stress, DNA-damaging agents, cytokines through various receptors, including tyrosine kinase receptors, and to a lesser extent by some G-protein-coupled receptors. 58 JNKs are activated by the Rho family of small GTPases. These proteins in turn activate MEKK1/4, MKK4/7, and JNK1/2/3. In addition to MEKK1/4, apoptosis signal-regulated kinase (ASK1), mixed lineage kinases (MLK1/3), and TGF β-activated kinase (TAK1) regulate the JNK pathway. 59 JNK pathway has been shown to play important roles in the control of cell proliferation, differentiation, apoptosis, and inflammation. 60 p38 MAPK was first identified in Saccharomyces cerevisiae as a protein kinase activated by hyperosmolarity, Hogl. 61 There are four isoformes of p38 MAPKs (α, β, γ, and δ) encoded from different genes. 48 Different isoformes are activated by inflammatory cytokines and various environmental stresses such as oxidative stress, UV radiation, hypoxia, ischemia, and others. Similar to JNKs, activation of p38 MAPKs through either stress or cell surface receptors involves members of the Rho family, which can activate and phosphorylate, MLKs, TAK1, ASK1, and MKK3/6. 48 In turn, MKK3/6 activates the four p38 isoformes. p38 pathway plays a critical role in normal immune and inflammatory responses, apoptosis, cell proliferation, and even survival. 62 The ERK5 pathway is one of the lesser studied and understood members of MAPK family. ERK5, also known as big MAPK (BMK1) because it is twice the size of other MAPKs, was initially found to be activated by oxidative stress and hyperosmolarity. 63 Subsequently, it was shown that ERK5 can be activated in response to serum, several growth factors, cytokines, and stress stimuli (reviewed by Drew et al). 64 The ERK5 signaling acts through sequential phosphorylation and activation of MEKK2/3, MEK5, and ERK5. The mechanism of activation of this pathway is still poorly elucidated; however, it is believed that several adaptor/scaffold proteins are involved, such as Lck-associated adapter 65 and Src. 66 ERK5 has been implicated in cell survival, differentiation, proliferation, and motility. In addition, several studies have suggested that ERK5 is involved in angiogenesis67,68 and may potentially regulate VEGF-mediated neovascularization. 69
AMD and MAPK Signaling
MAPKs have been implicated in many human pathologies, including neurodegenerative diseases (Alzheimer's, Parkinson's, and amyotrophic lateral sclerosis), diabetes, obesity, and different cancers. Given their pivotal role in key cellular processes, it is not surprising that alteration in expression and/or function of various intermediates of MAPK signaling is involved in the pathogenesis of AMD. Oxidative stress plays a central role in AMD. Commonly used experimental model to study the link between oxidative stress and AMD involves the use of cultured human RPE (ARPE19) cells. UV-induced damage is known to play a crucial role in eye diseases, including retinal degeneration. Studies have demonstrated that MAPKs ERK1/2, JNK, and p38 are activated in human RPE cells after UV exposure.23,70 A recent study demonstrated the protective effect of resveratrol on RPE cells against UV-induced damages through inhibition of MAPK activation. 71 Based on these results, it is suggested that resveratrol may act as a suppressing agent for prevention of UV-induced ocular disorders. 71 Furthermore, RPE cells exposed to the oxidant tert-butyl hydroperoxide (t-BHP) showed activation of ERK1/2 and several downstream nuclear targets. 19 Inhibition of the ERK pathway was found to completely block t-BHP-induced apoptosis, while neither JNK nor p38 MAPK inhibition was able to prevent the t-BHP-induced apoptosis in RPE cells. ERK was postulated as a potential target for treating oxidative stress-induced RPE degeneration, such as AMD. 19 Cadmium, released from cigarette smoke and metal industrial activities, caused activation of ERK, JNK, and p38 MAPK in ARPE19 cells, suggesting that cadmium could be an important factor in RPE cell death. 72 Studies from this laboratory and others have shown that constitutive activation of ERK by cadmium induces authophagic cell death. 73 Autophagy, or self-eating, is a catabolic process by which cells degrade and recycle cellular components in order to maintain cellular metabolism and homeostasis. 74 There is now considerable evidence that autophagy plays a significant role in RPE, and this process is less effective with aging. 75 On the other hand, it was demonstrated that Ras/Raf/MEK/ERK signaling pathway is involved in serum-induced RPE cell proliferation. 76 After hydrogen peroxide stimulation, ERK1/2 does not seem to be involved in cell death, but to be associated with oxidative stress-induced proliferation, 77 while JNK and p38 activation is important for hydrogen peroxide-induced apoptosis in RPE cells. 78 Short-term activation of ERK1/2 is associated with cell proliferation, 79 whereas persistent activation of ERK1/2 leads to cell death. 80
Many efforts have been made to establish animal models that mimic human AMD. The mouse is a genetically well-defined species and most of the retinal degeneration genes found first in mouse have been linked to a corresponding human retinal disease. The main drawback of using mice to study AMD is that the mouse retina has no macula. However, pathologic events seen in human AMD, such as drusen formation, thickened Bruch's membrane, various types of retinal degeneration, and CNV, have all been seen in various mouse strains. 81 It has been shown that deficient expression of the RNase III DICER1 leads to the accumulation of Alu RNA RPE of human eyes with geographic atrophy. 82 Studies revealed that Alu RNA overexpression or DICER1 knockdown increases the phosphorylation of ERK1/2 in mouse RPE in vivo, while JNK1/2 or p38 MAPK phosphorylation levels were unchanged. 26 Similar results were obtained when antisense oligonucleotide-mediated knockdown of DICER1 was used in primary human RPE cells. 26 PD98059, a potent and selective inhibitor of MEK, inhibits ERK1/2 phosphorylation and blocks RPE degeneration both in RPE cell culture and mice. 26 In addition, ERK1/2 pathway is also involved in experimental models of retinal and CNV.83,84 In a specific AMD model and in particular for retinal angiomatous proliferation, a form of wet AMD, the very low-density lipoprotein receptor knockout mouse (vldlr-/-), we have reported that ERK1/2, JNK, and p38 are elevated and a single intravitreal injection of cerium oxide nanoparticles (nanoceria) for one week reduces the phosphorylation of the three MAP kinases to the control levels. 28 A recent study demonstrates that JNK1 deficiency or JNK inhibition leads to a decrease in apoptosis, VEGF expression, and reduction of CNV in a mouse model of wet AMD (laser-induced CNV). 27
Using data from a whole-genome genotyping microarray and a multilocus enrichment analysis, SanGiovanni and Lee have identified AMD-associated regions related to an ∼30% change in the likelihood of having advanced AMD within six of 25 tested genes of JNK signaling pathway. 29 Makarev et al developed a new software AMD Medicine to evaluate the changes in functional pathway networks using changes in gene expression of single genes between AMD and normal eyes. 30 They discovered that several pathways including ERK are activated in RPE-choroid AMD phenotypes. The aforementioned findings indicate that MAPK pathways are undoubtedly involved in AMD pathology and are potentially attractive targets for both neovascular and atrophic AMD.
There is a crosstalk between MAPK signaling and several different pathways, such as VEGF, inflammasome, autophagy, and others. VEGF is known to be the major angiogenic factor involved in AMD.39–43 MAPK pathways play an essential role in modulating VEGF. It has been demonstrated that constitutive activation of ERK1/2 leads to increased VEGF expression, 85 and overexpression of p38 and JNK leads to elevated VEGF expression. 86 Inflammasomes play a central role in innate immune system. ROS serves as an important inflammasome signal that activates MAPK signaling. 87 It has been shown that dysregulation of inflammasome plays a role in various pathological conditions including AMD. 88 It was reported that NLRP3 inflammasome activation by drusen induces interleukin-18 (IL-18) secretion, which, in turn downregulates VEGF, thus reducing the excessive neoangiogenesis associated with AMD. 89 Zhang et al demonstrated that the cytokine IL-17A induced the activation of ERK1/2 and p38 MAPK in RPE cells. 90 Figure 2 shows a proposed model of MAPK signaling that leads to AMD.
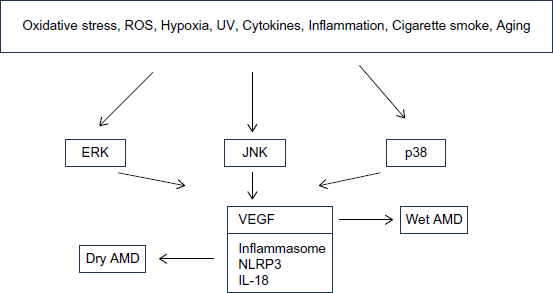
Schematic presentation depicting the role of ERK, JNK, and p38 in dry and wet AMD.
MAPK Inhibitors
Although the application of anti-VEGF drugs for treatment of wet AMD has significantly improved the control of disease, not all patients benefited from these drugs. Undoubtedly, identifying additional or alternative therapies that can improve the current standard treatment is of great interest.
MAPKs have become the most intensively studied protein kinases in the past two decades with a number of pharmacological inhibitors developed to block MAPK signaling. Dudley et al discovered the first small-molecule inhibitor of MEK1/2, the PD98059 compound. 91 Subsequently, another potent inhibitor of MEK1/2, U0126, was identified. 92 However, because of their pharmaceutical limitations, none of these two compounds have moved to clinical trials. However, PD98059 and U0126 have proven to be invaluable research tools to investigate the role of the ERK1/2 pathway in normal cell physiology and disease process. To date, several MEK1/2 inhibitors have been tested clinically or undergoing clinical trials for treatment of various cancers.93,94 These include trametinib (GSK1120212) for BRAF-mutated melanoma, selumetinib (AZD6244) for non-small cell lung cancer, binimetinib (MEK162, ARRY-162) for biliary tract cancer and melanoma, and PD0325901 for breast cancer, colon cancer, and melanoma. Raf inhibitors including vemurafenib, sorafenib, regorafenib, encorafenib (LGX818), and dabrafenib are in clinical trials for patients with tumor types harboring frequent mutations in BRAF gene and for treating renal, hepatocellular, and thyroid cancers. 95 However, adverse drug reactions including ophthalmologic complications occurred in patients treated with some MAPK inhibitors. For example, the incidence of retinal vein occlusion and retinal pigment epithelial detachments in patients treated with trametinib in clinical trials is 0.2% and 0.8%, respectively. 96 Uveitis occurred in 1% of patients receiving dabrafenib 97 and in 2.1% of patients treated with vemurafenib. 98 Therefore, these MAPK inhibitors cannot be used for treatment of AMD because of their ocular toxicity. The two broad spectrum inhibitors sorafenib and regorafenib are the most attractive drugs to target MAPK signaling in AMD. Both inhibitors target multiple kinases, including Raf, VEGF receptors 1–3, fibroblast growth factor receptor 1, and platelet-derived growth factor receptor, thereby inhibiting tumor growth and angiogenesis.99,100 No ocular toxicities were reported for sorafenib except one case of retinal tear possibly associated with the use of this drug. 101 Regorafenib (Stivarga; Bayer HealthCare) eye drops have been developed to inhibit VEGF activity in a small group of patients with neovascular (wet) AMD and a phase II trial has been recently completed (ClinicalTrials.gov Identifier: NCT02222207), pending results to evaluate the safety and tolerability of these eye drops. Because regorafenib is a multikinase inhibitor that inhibits VEGF, to what extent the inhibition of Raf/MEK/ERK signaling contributes to the clinical activity of this inhibitor is yet to be determined. Further understanding of the effects of sorafenib and regorafenib to target MAPK pathways in AMD is an area for research exploration. According to two new studies, presented as posters at the Association for Research in Vision and Ophthalmology 2015 Annual Meeting, regorafenib showed positive results as a potential topical therapy in the nonhuman primate laser-induced CNV model and in two different rodent models of ocular neovascularization.102,103 Because currently available treatments require intravitreal injections, regorafenib eye drops offer an innovative and noninvasive potential treatment option for wet AMD. However, local side effects are possible for theoretically every drug used in ophthalmology, which include toxicity related to the compound itself.
Additional pharmacological inhibitors that target p38 MAPK have been one of the most intensively studied classes of therapies for the treatment of inflammation. P38 MAPK inhibitors that are in phase II clinical trials for patients with autoimmune diseases and inflammatory processes include pamapimod, losmapimod, dilmapimod, doramapimod, VF-702, BMS-582949, ARRY-797, and PH-797804. 104 No ocular toxicities have been associated with the use of these inhibitors. Research into the role of p38 in AMD is an exciting area in the future that will be important to determine how best to make use of the therapeutic potential of targeting this signaling pathway.
Although there are several small-molecule JNK inhibitors such as SP600125, JNK-IN-8, and tanzisertib (CC-930), none of them proved to be effective in human tests yet.
Conclusion
In recent years, we have witnessed a mounting body of data suggesting that MAPKs are implicated in the pathogenesis of many human diseases. The evidence presented in this review indicates that MAPK pathways are involved in the development of AMD. Given the role of MAPK signaling in key cellular processes, interest in protein kinases as drug targets has exploded in the past few years. This may shift current research and clinical practice toward the use of MAPK inhibitors, alone or in combination with other therapeutics for treatment of AMD.
Author Contributions
Conceived the concepts: SVK. Analyzed the data: SVK. Wrote the first draft of the manuscript: SVK. Developed the structure and arguments for the paper: SVK. Made critical revisions: SVK. The author reviewed and approved the final manuscript.