
Abstract
Using Streptococcus pyogenes as a model, we previously established a stepwise computational workflow to effectively identify species-specific DNA signatures that could be used as PCR primer sets to detect target bacteria with high specificity and sensitivity. In this study, we extended the workflow for the rapid development of PCR assays targeting Enterococcus faecalis, Enterococcus faecium, Clostridium perfringens, Clostridium difficile, Clostridium tetani, and Staphylococcus aureus, which are of safety concern for human tissue intended for transplantation. Twenty-one primer sets that had sensitivity of detecting 5–50 fg DNA from target bacteria with high specificity were selected. These selected primer sets can be used in a PCR array for detecting target bacteria with high sensitivity and specificity. The workflow could be widely applicable for the rapid development of PCR-based assays for a wide range of target bacteria, including those of biothreat agents.
Introduction
The rapid and economic detection, identification, and quantification of infectious agents in various clinical settings are essential. Currently, the “gold standards” for the detection of bacterial and fungal agents have been the traditional microbiological culturing methods that are generally laborious and time-consuming.1,2 Therefore, it is highly desirable to develop alternative molecular methods, such as nucleic acid-based tests (NATs), which have exquisite sensitivity and a relatively short turnaround time.
Recent advances in detection technology have produced several new systems that allow the detection of a wide range of pathogens. For example, TessArae uses high-density and high-resolution microarrays for microbial identification based on resequencing of selected target genes by hybridization. 3 Taking advantage of the high accuracy of Matrix-assisted laser desorption/ionization (MALDI) to measure the mass of DNA fragments with high precision, PLEX-ID utilizes the base composition of specific DNA fragments measured by MALDI for microbial identification.4–6 The capacity for these two systems to detect a wide range of pathogens, from viruses and bacteria to protozoa, is very high. However, additional processing time is required for hybridization and MALDI for TessArae and PLEX-ID, respectively, and the results are in general not quantitative. In comparison, real-time PCR assays are more desirable for rapid detection of the targeted or selected pathogens. Multiple primer sets can be arranged in an array format on a 96- or 384-well plate for multiplex detection.
In developing any NATs, it is most critical to effectively select “DNA signatures” (ie, species-specific sequences) from the target microbe of interest for highly sensitive and specific assays. 7 DNA signatures should be highly conserved within the target species and absent in any other species of microorganisms, including those of closely related microbial species. In spite of the importance of identifying DNA signatures in assay development, the processes for selecting DNA signatures have generally not been well described in the literature.8,9
In our previous study, we established a computational workflow using Insignia, dCAS, and NCBI-BlastN sequentially for the identification of DNA signatures from whole genome sequence data that can be used as the basis for designing real-time PCR assays using Streptococcus pyogenes as a model organism. 10 In this study, we extended the use of this approach for rapid development of PCR tests against six additional bacterial pathogens. Enterococcus faecalis, Enterococcus faecium, Staphylococcus aureus, Clostridium perfringens, Clostridium difficile, and Clostridium tetani have been defined as “high-risk” bacteria American Association of Tissue Banks (AATB) in tissue grafts. 11 It is our final objective that a PCR array can be developed for multiplex detection of all the high-risk bacteria pathogens in testing during and after tissue processing.
Materials and Methods
Microbial strains
Microbial strain E. faecalis (ATCC 29212), E. faecium (ATCC 35667), S. aureus (ATCC 25923), C. perfringens (ATCC 3628, ATCC 13124), and C. difficile (ATCC 17857, ATCC 43255) were obtained from American Type Cell Collection (ATCC). C. tetani strain Massachusetts C2 was kindly provided by James Keller (FDA). Microorganisms were cultured following protocols recommended by ATCC. The identities of these microbial strains were confirmed by Biolog Microbial Identification System GEN III (Biolog), which utilizes metabolic characteristics such as pH, salt, lactic acid tolerance, reducing power, chemical sensitivity, and the ability of the cell to metabolize all the major classes of biochemicals to identify microbial species.
Isolation of genomic DNA
To isolate genomic DNA from microorganisms, cell pellets were harvested from either broth or solid cultures, and genomic DNAs were isolated using the DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer's protocol. Human genomic DNA was prepared from buffy coat cells of a healthy blood donor from NIH Blood Bank using the DNeasy Blood & Tissue Kit or the conventional phenol/chloroform extraction method. DNA was quantified using Nanodrop (Thermo Scientific) and stored at −20°C prior to PCR amplification.
Identification of final DNA signatures and design of PCR primer sets
The overall computational workflow has been previously described. 10 Briefly, the open-internet-accessible Insignia program 12 , 13 (http://insignia.cbcb.umd.edu) was selected to produce DNA signatures for E. faecalis, E. faecium, S. aureus, C. perfringens, C. difficile, and C. tetani. Currently, there are a total of 13,928 genome sequences in Insignia's database, with 11,274 from virus/phages and 2,653 from nonvirus organisms. To identify DNA signatures for a target species, the Insignia program first calculated conserved regions within genomes of all available strains of the target species. Subsequently, DNA signatures were generated by excluding shared regions with all nontarget (background) genomes and output in FASTA format using the genomic coordinates of the reference genome as their description. However, we realized that the majority of these initial DNA signatures had high homology with sequences of closely related species. Therefore, it was necessary to use the dCAS program 14 as a second screening to select the top 50 DNA signatures that were more different from the sequences of closely related species. The specificity of the top 50 DNA signatures was then subjected to the third screening against the NCBI nonredundant database to select for final DNA signatures that do not have high homology (generally E-value > 1, bit score < 40) with any other known DNA sequences by NCBI-BlastN. 15 All oligonucleotide primer sets were then designed for these final DNA signatures using Primer-Blast (http://www.ncbi.nlm.nih.gov/tools/primer-blast/), 16 and named after the target species (Efl, Efm, Cp, Cd, Ct, and Sa for E. faecalis, E. faecium, C. perfringens, C. difficile, C. tetani, and S. aureus, respectively). Since the PCR assays will be used in the presence of human DNA, primer set specificity was checked to ensure that no likely theoretical amplification from human DNA was predicted.
Conventional and real-time PCR testing and melting curve analysis
To perform conventional PCR assays, AmpliTaq gold DNA polymerase (Applied Biosystems) was used. PCRs (20 μL) were performed in 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 2 mM MgCl2, 200 μM dNTPs, 50 pmol of each primer with 1 U AmpliTaq gold DNA polymerase (Applied Biosystems) using Eppendorf Mastercycler ep gradient S (Eppendorf) with the following parameters: an initial activation of AmpliTaq gold DNA polymerase at 94°C for ten minutes, followed by 45 cycles of 94°C for 30 seconds (denaturation), 55°C for 30 seconds (annealing), and 72°C for 30 seconds (extension), followed by a final extension at 72°C for seven minutes. Amplicons were separated on 2% agarose gel by electrophoresis, stained with ethidium bromide, and detected upon UV transillumination by an electronic documentation system (GelDoc-It Imaging System, UVP).
To perform real-time PCR assays, Power SYBR® Green PCR Master Mix with AmpliTaq gold DNA polymerase was used. Real-time PCR was performed in triplicates on CFX96 real-time PCR Detection System (Bio-Rad) with the following parameters: an initial activation of AmpliTaq gold DNA polymerase at 94°C for ten minutes, followed by 50 cycles of 94°C for 15 seconds (denaturation) and 60°C for 60 seconds (annealing and extension). Amplicons were subjected to melting curve analysis by increasing temperature from 65°C to 95°C at 0.5°C per second, recording the changes in fluorescence to determine the melting profiles for each detected signal. The threshold of each PCR reaction is determined by the CFX manager software (Bio-Rad) with default setting.
Results
After the size adjustment, the number of DNA signatures remaining for E. faecalis, E. faecium, C. perfringens, C. difficile, C. tetani, and S. aureus, were 844, 278, 237, 583, 3,322, and 913, respectively. As a second screening, each species' DNA signatures were then compared to a local database containing the genome sequences of their respective closely related species within the same genus by dCAS. Then, the top 50 DNA signatures with highest E-value compared to the closely related species, indicating their sequence distinctness, were selected for a third screening against the NCBI nonredundant database by BlastN. A total of 5, 11, 10, 4, 3, and 10 final DNA signatures were selected for primer design for E. faecalis, E. faecium, C. perfringens, C. difficile, C. tetani, and S. aureus, respectively. Table 1 lists the locations of these DNA signatures, based on the genomic coordinates of the type strain, and their annotated gene information.
Evaluation of primer sets designed from selected signature sequences of E. faecalis (Efl), E. faecium (Efm), C. prefringens (Cp), C. difficile (Cd), C. tetani (Ct) and S. aureus (Sa) by 2 PCR platforms.
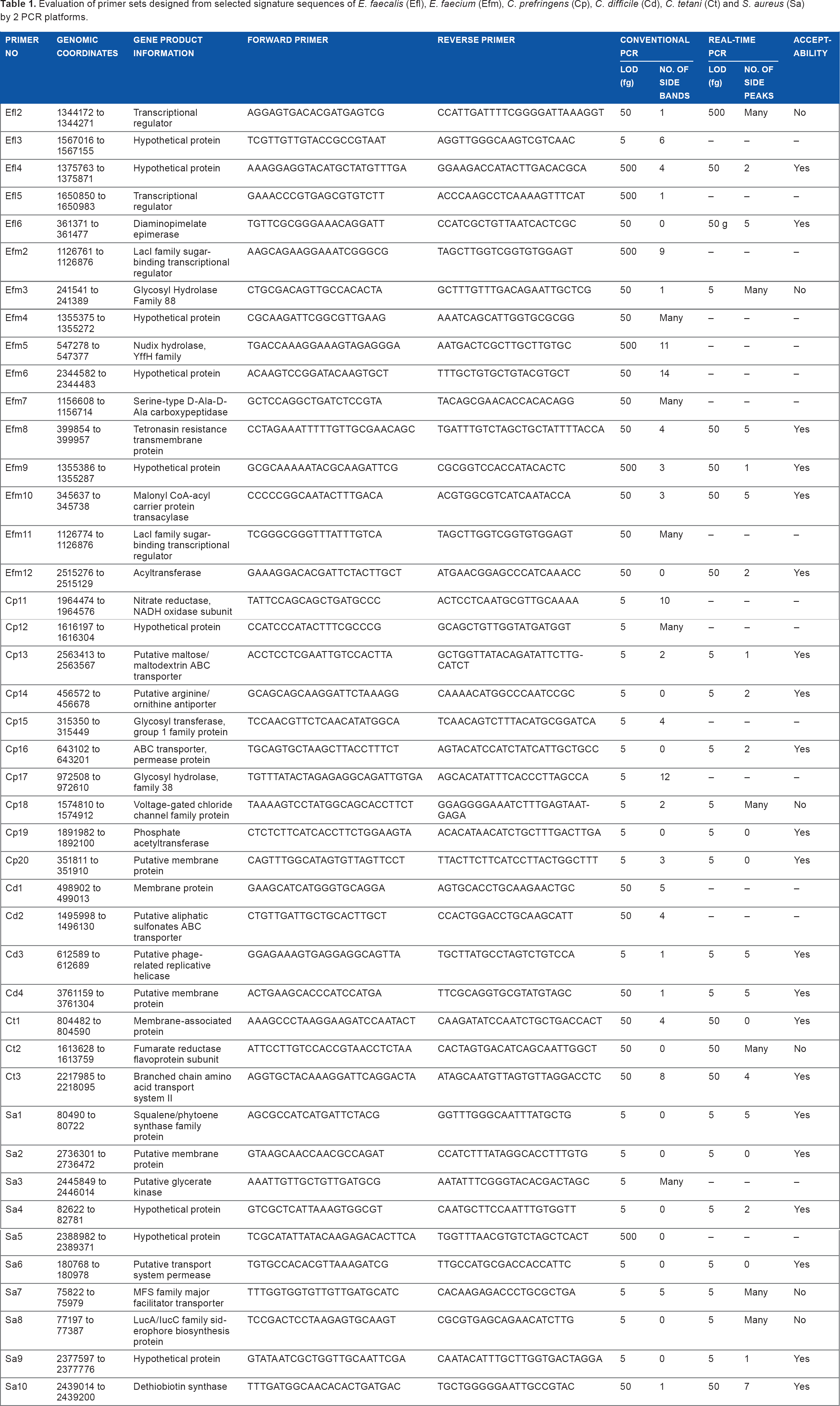
The Primer-BLAST program was used to design primer sets based on the final selected DNA signatures. The optimal melting temperature (Tm) for all primers was set at 55°C with a maximum Tm difference between the forward and reverse primers of 3°C, and the length of amplicons was generally limited to 150 bp for maximum PCR efficiency. Table 1 lists the sequences of all primer sets.
Assessment of the sensitivity and specificity of species-specific primer sets in conventional PCR assays
Following the theoretical design of 43 primer sets in silico, we first evaluated the sensitivity and specificity of these primer sets using conventional PCR assays. A typical testing comprised a panel of DNA samples consisting of 5 pg, 500 fg, 50 fg, and 5 fg of DNA from each target pathogen with and without the presence of 5 ng human DNA, and 5 ng of DNA from 39 bacterial species, including closely related species, other common Gram-positive and Gram-negative bacteria, 5 ng of DNA from three common human yeast pathogens Candida albicans, Candida tropicalis, and Candida parapsilosis, and 5 ng of human DNA alone were included in the testing. A representative conventional PCR testing of primer sets Efl6, Efm12, Cp14, Cd3, Ct3, and Sa2 is shown in Figure 1. Specific amplicons of the expected sizes from as low as 5–50 fg of DNA from their respective target species were produced. In most cases, no amplicons of the expected sizes were produced using 5 ng DNA (a million-fold excess) of nontarget species (Fig. 1 and Table 1). Among the 43 selected primer sets, 20 primer sets (Efl3, Cp11–Cp20, Cd3, Sa1–Sa4, Sa6–Sa9) detected 5 fg DNA of the respective target species; 17 primer sets (Elf2, Efl6, Efm3, Efm4, Efm6–Efm8, Efm10–Efm12, Cd1, Cd2, Cd4, Ct1–Ct3, and Sa10) detected 50 fg DNA of the respective target species; and six primer sets (Elf4, Efl5, Efm2, Efm5, Efm9, and Sa5) detected 500 fg DNA of the respective target species (Table 1). In most cases, the limit of detection was not affected by the presence of 5 ng human DNA in the background.

Representative conventional PCR testing of species-specific primer sets (Elf6, Efm12, Cp14, Cd3, Ct3, and Sa2) using 5 pg, 500 fg, 50 fg, and 5 fg of purified target genomic DNA (lanes 1–4 without human DNA and lanes 5–8 with 5 ng of human DNA, respectively, for E. faecalis, E. faecium, C. clostridium, C. difficile, and C. tetani; lanes 45–48 without human DNA and lanes 49–52 with 5 ng of human DNA for S. aureus) and 5 ng of purified DNA from respective nontarget species: S. pyogenes, Streptococcus dysgalactiae group G, Streptococcus agalactiae group B, Streptococcus dysgalactiae group C, Streptococcus sp. strain H60R group F, Streptococcus viridans, Streptococcus equi Group C, Streptococcus gallolyticus, Streptococcus mutans, Streptococcus uberis, Streptococcus salivarius, Streptococcus thermophilus, Streptococcus pneumoniae, Staphylococcus epidermis, Listeria monocytogenes, Corynebacterium sp., Bacillus subtilis, Bacillus cereus, Bacillus circulans, Bacillus polymyxa, Bacillus megaterium, Bacillus sphaericus, Bacillus thuringiensis, Bacillus coagulans, Bacillus alvei, Escherichia coli, Salmonella typhimurium, Salmonella sp. group D, Pseudomonas fluorescens, Shigella sonnei, Enterobacter aerogenes, Klebsiella pneumoniae, Morganella morganii, Proteus mirabilis, Alcaligenes odorans, Stenotrophomonas maltophilia, C. albicans, C. tropicalis, C. parapsilosis, human being, and a no template control.
Assessment of the sensitivity and specificity of selected species-specific primer sets in real-time PCR assays. To avoid potential problems with data interpretation, we eliminated 16 primer sets from a total of 43 primer sets (denoted as “–” in Table 1) from the real-time PCR testing: those that had higher limits of detection; those with too many side products amplified from nontarget species; or those that had nontarget amplicons close to those of the target species. A representative real-time PCR testing of primer sets Efl6, Efm12, Cp14, Cd3, Ct3, and Sa2 is shown in Figure 2. Among the remaining 27 primer sets, 22 primer sets had the same sensitivity as the conventional PCR assay, which detected 5–50 fg target DNA (Table 1). We noted with interest that the assay sensitivity for primer sets Efl4, Efm3, Efm9, and Cd4 was improved in real-time PCR testing, compared to the conventional PCR testing. On the contrary, primer set Efl2 was less sensitive in real-time PCR testing (Table 1).

Representative real-time PCR testing conducted with species-specific primer sets (Elf6, Efm12, Cp14, Cd3, Ct3, and Sa2) using 5 pg (blue), 500 fg (orange), 50 fg (pink), and 5 fg (black) of target genomic DNA, and 5 ng of purified DNA from respective nontarget species: S. pyogenes, Streptococcus dysgalactiae group g, Streptococcus agalactiae group B, S. dysgalactiae group c, Streptococcus sp. strain H60R group F, Streptococcus viridans, Streptococcus equi Group C, Streptococcus gallolyticus, Streptococcus mutans, Streptococcus uberis, Streptococcus salivarius, Streptococcus thermophilus, Streptococcus pneumoniae, Staphylococcus epidermis, Listeria monocytogenes, Corynebacterium sp., Bacillus subtilis, Bacillus cereus, Bacillus circulans, Bacillus polymyxa, Bacillus megaterium, Bacillus sphaericus, Bacillus thuringiensis, Bacillus coagulans, Bacillus alvei, Escherichia coli, Salmonella typhimurium, Salmonella sp. group D, Pseudomonas fluorescens, Shigella sonnei, Enterobacter aerogenes, Klebsiella pneumoniae, Morganella morganii, Proteus mirabilis, Alcaligenes odorans, Stenotrophomonas maltophilia, C. albicans, C. tropicalis, C. parapsilosis, human being, and a no template control (green).
With regard to the assay specificity, primer sets Efl2, Efm3, Cp18, Ct2, Sa7, and Sa8 produced many side amplicons from nontarget DNA with similar melting peaks close to those of the respective target species, and thus were considered “unacceptable” (Table 1). In comparison, four primer sets (Cp19, Cp20, Ct1, and Sa6) showed very high specificity, with no above-threshold amplification observed from all their respective nontarget DNA tested (Table 1 and Fig. 2). The remaining 17 primer sets were also considered “acceptable” (Table 1) as the side peaks produced from their respective nontarget DNA using each of these primer sets are clearly distinguishable from their respective target DNA, based on their distinct melting profiles. These 21 acceptable primer sets will be selected for future study.
Discussion
It is highly advantageous to develop nucleic acid-based molecular methods with both high sensitivity and specificity for rapid detection of pathogenic microbes. However, the true challenge to develop such molecular assays critically depends on the effective identification of truly species-specific DNA signatures as primers for PCR. In order to meet the critical challenge, we previously developed a stepwise computational workflow to effectively identify highly species-specific DNA sequences that could be used to detect the target bacteria in high sensitivity using S. pyogenes as a model organism. 10 In this study, we further tested the computational workflow for its effectiveness to identify species-specific DNA sequences intended as primers to be used in real-time PCR assays for the rapid detection of six different bacterial pathogens.
In the initial step of the workflow, tens of thousands of DNA signatures of 20–385 bp in length were identified by Insignia program for each pathogen studied. However, our previous study of S. pyogenes revealed that PCR primers designed from many of these signatures would still amplify DNA from many closely related species of bacteria. The Insignia algorithm identified as few as one difference out of every 20 nucleotides against all background genomes as DNA signature. A valid DNA signature could have up to 95% similarity in sequence with its close relatives. We used dCAS program to effectively differentiate the degree of sequence homology between the DNA signatures produced by Insignia program and the genomic sequences of closely related species by the E-values of their sequence alignments. The top 50 DNA signatures for each of the six pathogens were selected for final verification of sequence uniqueness by BlastN against NCBI nonredundant nucleotide database. Using the workflow, we selected 43 candidates of DNA signatures (>102 bp) that could be used for designing primers (Table 1) and experimentally evaluated them for detection sensitivity against the target bacteria and specificity of not amplifying a panel of nontarget bacteria at a concentration of a million folds higher than those of target bacteria using both conventional PCR testing and real-time PCR testing.
A recent study by Zhang and Sun 17 described successful identification of tens of thousands of uniquely conserved regions (UCRs) of 18 nucleotides from bacteria genome using in-house bioinformatic pipeline. The study similarly showed that the UCRs identified could be used as primer sets for the detection of 15 target bacterial species. Both Insignia program and UCRs pipeline utilize the principle of k-mer. Insignia program relies on the precomputed pairwise comparison database, whereas UCRs pipeline converts nucleotide sequence into numerical value using integer mapping methods for the identification of species-specific sequences. In the study of UCRs pipeline, the limits of detection for target bacteria by the eight primer sets generating amplicons of 576–1110 bp in size were not specifically examined. In our study, the limit of detection of the final 21 selected primer sets for the six pathogens (Table 1) ranged from 5 to 50 fg (equivalent to 1–10 genomic copies) per reaction, which is equivalent to previously reported assays with high sensitivity. 9 We believe the high sensitivity observed in our PCR assays could in part be attributed to the sizes of amplicons produced by the selected DNA signatures that were less than 150 bp. Importantly, the high sensitivity of detection against the target bacteria using these selected primer sets were achieved at the same time with extremely high specificity of the assays. None of the 21 primer sets selected would amplify human DNA and a panel of DNA from 42 nontarget species of bacteria tested at a concentration of a million folds higher than those of the target bacteria.
The target sequences for the DNA signatures we identified and studied for these bacteria were different from those used in various studies previously reported,8,18,19 most of which selected target genes based on their species-specific biochemical or immunological properties. It is unclear how well the selected target gene sequences are conserved among different strains of the same species of bacteria by this approach, which is evidently different from our computational workflow using whole genome sequences to search for DNA signatures conserved among multiple strains within the target species. The rapid advance of the next generation of sequencing technology has already made many more microbial genome sequences available in the public domain. It is important to note that as more new genome sequences become available,20,21 the currently validated DNA signatures should be reexamined against the new genome sequences to ensure the conservedness. Our approach or protocol of finding species-specific DNA signature should become much more stringent in the future.
In conclusion, we demonstrated that our computational workflow effectively provided a simple genome-wide approach to rapidly identify multiple, highly specific primer sets against six target pathogens. We evaluated individual primer sets under the same condition in real-time PCR assays, these primers and reagents could be easily assembled into a 96- or 384-well plate as a PCR array for multiplex detection of these target bacteria pathogens. Since the capacity of PCR array is high, more validated primer sets for additional pathogens designed in the future could be quickly incorporated for simultaneous detection of an extended list of pathogens of interest. The approach described here could prove highly valuable in the rapid development of a highly sensitive and specific conventional or real-time PCR assay for the detection of any target pathogen, including those of biothreat agents.
Footnotes
Acknowledgments
The Core Facility of CBER, FDA provided oligonucleotide synthesis. We acknowledge Drs. Adam Phillippy and Jason Guo for their technical assistance with the Insignia and dCAS programs, respectively. We also thank Drs. Shien Tsai, Brenton McCright, and Prajakta Varadkar for critically reading the manuscript. The FDA Library's editorial service is also acknowledged.
Author Contributions
Conceived and designed the experiments: GCH and SCL. Analyzed the data: KN, GCH, BL and SCL. Wrote the first draft of the manuscript: GCH. Contributed to the writing of the manuscript: KN, GCH, BL and SCL. Agree with manuscript results and conclusions: KN, GCH, BL and SCL. Made critical revisions and approved final version: GCH and SCL. All authors reviewed and approved of the final manuscript.