
Abstract
A relationship between obesity and type 2 diabetes is now generally well accepted. This relationship represents several major health hazards including morbid obesity and cardiovascular complications worldwide. Diabetes mellitus is a complex metabolic disorder characterized by impaired insulin release and insulin resistance. Lipids play an important physiological role in skeletal muscle, heart, liver and pancreas. Deregulation of fatty acid metabolism is the main culprit for developing insulin resistance and type 2 diabetes. A predominant predisposing factor to developing obesity, insulin resistance and type 2 diabetes is the permanent elevation of free fatty acids in plasma followed by impaired utilization of lipids by muscle. Diabetes-induced inflammation and oxidative stress have also vital role for development of insulin resistance in diabetic patients. The present review is intended to describe the correlation between lipids, obesity and insulin resistance based on current literature, in order to elucidate involved molecular mechanisms in depth.
Introduction
Type 2 diabetes is a complex metabolic disorder characterized by a persistent high rise in blood glucose along with the modification of some biochemical processes, including lipolysis, lipid peroxidation, and dearth of production of glucose transporters, resulting from defects in insufficient insulin secretion, inefficient insulin function or both. 1 Obesity is a condition in which there is excess storage of fat in the body leading to cardiovascular morbidities, insulin resistance and even the development of type 2 diabetes. In obese patients, adipose tissue releases a high amount of non-esterified fatty acids (NEFA), glycerol, hormones, and proinflammatory cytokines. These elements are involved in the development of insulin resistance. When insulin resistance is accompanied by dysfunction of pancreatic islet β-cells, the cells fail to control blood glucose level due to insulin release impairment. These abnormalities in β-cell function cause development of type 2 diabetes. High lipid level in blood has long been associated with type 2 diabetes. Patients with non-insulin-dependent diabetes mellitus (NIDDM), also known as type 2 diabetes mellitus, sometimes have abnormal lipid profiles. Type 2 diabetes is one of the free radical diseases, which increases complications with increased lipid peroxides. 2 In this disease, oxidative stress is increased due to an increased production of oxygen-free radicals and inefficient performance of the antioxidant defense system. 3 Increased oxygen-free radicals result in the lipid peroxidation of cellular lipids and play a vital role in atherosclerosis and microvascular complications in type 2 diabetes. 4 Type 2 diabetes is related to significant cardiovascular morbidity and mortality by modulation of lipid profiles. Dyslipidemia, which occurs in approximately 50% of patients with type 2 diabetes, results in cardiovascular complications by elevated triglyceride (TG) levels, low high-density lipoprotein (HDL) cholesterol levels, and high rise of small, dense low-density lipoprotein (LDL) cholesterol. 5 Dyslipidemia, therefore, may be characterized by raised small, dense LDL levels, elevated levels of TG, and low levels of HDL. Individually, the latter two factors increase the risk of cardiovascular diseases, and the combination of these is a risk factor for cardiovascular complications. This risk factor is at least as strong as that of having a high level of LDL cholesterol. 6
Insulin resistance is a condition in which cells do not respond to the normal action of insulin secreted from the pancreatic β-cells. As a result, cellular uptake of glucose does not occur and blood glucose level is elevated. This leads, finally, to type 2 diabetes. Resistance to insulin has a direct link to the changes in lipid profiles in NIDDM, and usually it is associated with higher concentrations of TG, and lower concentrations of HDL. 7 Insulin resistance develops a number of alterations in lipid metabolism and lipoprotein composition, which renders LDL cholesterol and other lipoproteins more pathogenic in patients with type 2 diabetes. Hypertriglyceridemia results in decreased HDL, which is also a main feature of plasma lipid alterations observed in type 2 diabetic patients. 8 The low level of HDL, which exerts anti-atherogenic and antioxidative effects when present in sufficient amounts, is a key feature of NIDDM. Elevation in plasma TG level is observed when HDL level is reduced. This process is mediated by cholesterol ester transfer protein (CETP). Hypertriglyceridemia may be an increased hepatic secretion of very low-density lipoproteins (VLDL) and a delayed clearance of TG-rich lipoproteins, which may mainly be due to increased levels of substrates for TG production, and enhanced free fatty acids (FFA), and glucose levels. The latter could be secondary to decreased activity of lipoprotein lipase (LPL), a key enzyme for lipoprotein-TG. 9 Thus, there is a strong association between type 2 diabetes and dyslipidemia. The prevalence of obesity worldwide and its increasing association with diabetes demand for a mechanistic understanding of obesity-related insulin resistance as a priority to understand this correlation in a defined way. In this review we will focus particularly on molecular aspects by which lipids/FFA promote insulin resistance to elucidate the strong association between obesity, insulin resistance and type 2 diabetes.
Fatty Liver Disease, Obesity and Type 2 Diabetes
Non-alcoholic fatty liver disease (NAFLD) leads to obesity and type 2 diabetes.10,11 Hepatic TG content in NAFLD is directly related to the free fatty acid uptake by liver. FFA are released from subcutaneous adipocytes. FFA enter systemic circulation and are transported to the liver through the hepatic artery and portal vein. Additional FFA are released by lipolysis of visceral adipose tissue TG. 12 Free fatty acid release rate is higher in case of obese patients than lean. 13 Elevated hepatic lipase and hepatic lipoprotein lipase with postprandial lipemia and increased concentrations of FFA have been found in NAFLD patients, which may be responsible for the high concentration of postprandial incorporation of dietary fatty acids. 14 Intra-hepatic TG (IHTG) in obese patients is associated with type 2 diabetes. 15 Interestingly, liver also produces FFA by de novo synthesis through a series of complex cystosolic polymerization reactions. 16 Hepatic fatty acid de novo synthesis is regulated particularly in response to insulin and glucose. 16 Transcription factors, sterol regulatory element binding proteins or SREBPs and carbohydrate responding element binding proteins or ChREBPs are activated, which induce nearly all genes involved in de novo lipogenesis (DNL). 16 In cases of NAFLD in humans, hepatic expression of several genes involved in DNL additionally increase insulin resistance in skeletal muscles. This may promote IHTG accumulation by diverting ingested carbohydrate (from high carbohydrate meal) away from storage and lower de novo fatty acid synthesis. 17 DNL plays an important role to control total IHTG synthesis and VLDL-TG secretion is much higher in NAFLD patients in comparison to normal individuals. 16 After taking food, cholesterol is absorbed into gastrointestinal cells and gets mixed with nascent chylomicrons, hepatic uptake of which provides sources of FFA in the liver. Lipoproteins are mostly produced in the liver. Lipid molecules are always in plasma circulation as lipoprotein complexes. Triglycerides are stored in the hepatic tissue as lipid droplets, secreted and circulated in the form of VLDL in the blood. Then VLDL is converted to intermediate density lipoprotein (IDL), and subsequently to LDL with higher cholesterol content, by the action of LPL. In cases where excess energy is needed, glucose is converted to fatty acids, and then to TG. 6 After that, LDL circulates in the blood, reaches hepatocytes and binds with LDL receptor. In patients with insulin resistance, hepatic lipid homeostasis is maintained by the increased secretion of VLDL. The plasma NEFA-pool contributes the majority of the fatty acids that flow to the liver in the fasted state, which is the major source of fatty acids secreted by the liver in the form of VLDL. 18 Insulin resistance, therefore, has a direct association with elevated lipid and lipoprotein concentrations, elevated VLDL production, and increased plasma LDL level. 11 LDL molecules stay longer in the circulation, as LDL receptors show a lower affinity for these smaller particles. 19 By low activity of LPL, or by high level of apolipoprotein C-3 or APOC-3, an inhibitor of LPL, a high lipid level occurs in the blood. 9 As compared to steatosis, the steatohepatitis mostly links to abnormal VLDL synthesis and secretion. 20 Thus, liver has distinct role in the development of insulin resistance.
Role of FFA in the Development of Insulin Resistance
A rise in FFA concentration has important physiological consequences; for example, during pregnancy, an elevated level of FFA induces insulin resistance and so valuable glucose is conserved for the developing foetus. 21 Thus, FFA plays both a physiological and a pathological role in the body. But excess FFA in blood may cause serious metabolic syndrome. Over 80% of people with type 2 diabetes are obese and insulin resistant. 22 Obesity and insulin resistance may be linked with some mediators including FFA, tumor necrosis factor-α (TNF-α), leptin and adinopectin. Adipocytes are claimed to be a site of insulin resistance. 23 Insulin resistance increases lipolysis, leading to increased concentration of circulating FFA and to the development of insulin resistance in skeletal muscles and the liver. 24 Thus, there is a strong association between increased plasma FFA, intramyocellular lipid accumulation and insulin resistance. 22 Although there are many studies suggesting strong link of FFA and insulin resistance, the mechanism is not fully understood and more research is required to find the exact molecular basis.
Molecular Pathophysiology of Diabetes
For many years, scientists have been trying to understand pathophysiology at the molecular level to control the elevated blood glucose in type 2 diabetes. 25 Type 2 diabetes is reaching an epidemic stage with the increased prevalence of obese people. 26 The development of type 2 diabetes results from an interaction of a patient's genetic background 27 with social and environmental factors (Fig. 1). Disorders of the nervous and endocrine systems, a sedentary life-style, psychological stress, a high fat diet, overeating, age, smoking and alcohol intake are some of the causes which pave the way to obesity, leading to diabetes. Alteration in communication among some vital organs such as the liver, skeletal muscles, pancreas, fat tissue, gastrointestinal tract and brain may result in an elevated blood glucose level, leading to type 2 diabetes. Since most patients with type 2 diabetes are overweight or obese, the role of fatty acid cannot be ignored in the development of diabetes. An interaction of NEFA with glucose metabolism has been described. 28 The correlation between obesity and insulin resistance may be assumed to be a “cause and effect” relationship, since clinical and preclinical studies indicate that weight loss/gain correlates closely with increasing/ decreasing insulin sensitivity. 29 The major influence of fat distribution, especially the negative influence of intra-abdominal or visceral fat depot, is now largely established. 30 Hyperglycemia has been claimed to be a prerequisite for lipotoxicity to occur, and therefore the term glucolipotoxicity, rather than lipotoxicity, is more appropriate to describe the deleterious effects of lipids on β-cell function. 31 The role of obesity in the pathophysiology of type 2 diabetes and insulin resistance has been attested to in several studies.32,33 Although the cause of inflammation in obesity has not yet been elucidated fully, various metabolic and immune pathways are claimed to be closely involved. The role of immune cells in promoting inflammation in obesity has been confirmed in humans. 34 Obesity initially develops pro-inflammation starting from metabolic cells (adipocyte, hepatocyte, or myocyte) and eventually recruits immune cells with the release of inflammatory cytokines such as TNF-α, interleukin (IL)-6, and adiponectin. Secretion of leptin, TNF-α, resistin, adiponectin, inducible nitric oxide synthase (iNOS) and an elevated plasma NEFA level gradually leads to obesity-induced inflammation that may interfere with glucose metabolism and insulin sensitivity and produce type 2 diabetes. 35 The enzyme iNOS is a key inflammatory mediator in obesity and causes insulin resistance in the skeletal muscles. It inhibits secretion of adiponectin from adipocytes and impairs insulin secretion in the liver. Elevated iNOS in blood vessels causes vascular dysfunction in obesity. 36 Macrophage accumulation in adipose tissues in obese patients shares the expression of multiple genes causing adipose tissue inflammation in obesity. 37 Similarly, some genetic modifications such as the glucokinase gene, insulin receptor substrate-I (IRS-I), mitochondrial genes, and so on alter insulin secretion or function, leading to type 2 diabetes.
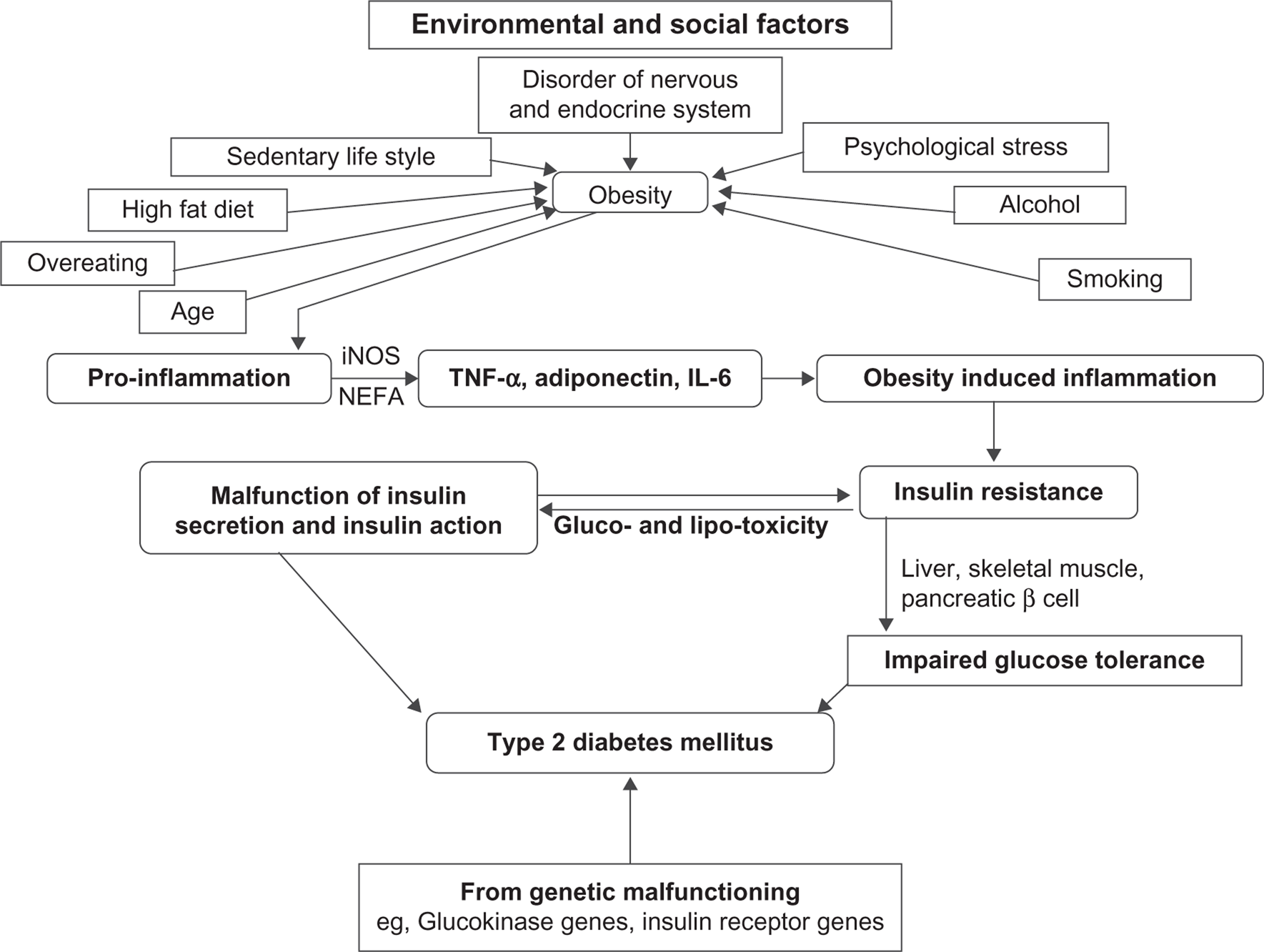
Combined impact of genetic makeup, environmental and social factors in the process of development of type 2 diabetes associated with obesity through impaired insulin secretion and insulin action, explaining the progression from insulin resistance to an impaired glucose tolerance test (IGT) and type 2 diabetes.
Obesity to Insulin Resistance in Type 2 Diabetes
Here we have projected three important pathways that are directly associated with the development of insulin resistance.
mTOR pathway promotes insulin resistance and type 2 diabetes by nutrient sensing
Excess nutrients have been reported to promote insulin resistance by activating the protein kinase mammalian target of the rapamycin (mTOR) pathway. 38 mTOR causes serine phosphorylation (pS) of IRS-I, by activating S6 Kinase1 (S6K1) and uncoupling IRS-I from the activation of phosphatidylinositol 3-kinase (PI3K) and Akt proteins, the target proteins of the metabolic pathway of insulin 39 (Fig. 2). This type of feedback has been found in myocytes, adipocytes and hepatocytes, 34 indicating that in the regulation of glucose homeostasis, the mTOR pathway plays a very important role. Over-expression of mTOR and its effector S6K1 in skeletal muscle, liver and adipose tissue has been reported in genetic and dietary animal models. 40 Serine 1101 in the IRS-I protein has been suggested to be a molecular target of S6K1 in the liver of obese animals during the administration of amino acids by infusion in skeletal muscles. 4 Adipocytes or macrophages in adipose tissue or skeletal muscle release various types of proinflammatory cytokines (TNF-α, IL-1 and IL-6). These mediators activate c-jun N-terminal kinase (JNK) and inhibitor kappa B kinase (IKK). IKK and JNK inhibit pS of IRS-I, a key element of the insulin-signaling cascade, through the transcriptional activation of inflammatory genes such as iNOS and the promotion of insulin resistance. iNOS activation leads to high levels of nitric oxide (NO) production, which in turn produces the free radical peroxynitrite (ONOO-). This free radical causes s-nitrosylation or nitration of IRS-I, PI3K and/or Akt, to provide inhibitory action on them. Activation of PI3K and Akt is indispensable for glucose transporter 4 (GLUT4) translocation to the cell surface. As stated earlier, when IRS-I is phosphorylated at the serine residue, due to uncoupling of IRS-I, the activation of PI3K and Akt proteins do not occur. Instead, this inhibits the membrane translocation of GLUT4 protein to transport glucose from outside of the cell to the inside. On the other hand, dephosphorylation of tyrosine residue deactivates the IRS-I protein. Excess nutrients cause insulin resistance by activating the mTOR /S6K1 pathway, which in turn inhibits proliferator–activated receptor– gamma coactivator-1 (PGC1) expression. Inhibition of PGC1 gene expression reduces mitochondrial energy expenditure, leading to obesity. On the other hand, excess nutrients reduce IRS tyrosine phosphorylation (pY) via hexosamine, the IKK/protein kinase C (PKC) pathway and ceramides signaling. Activation of adenosine monophosphate (AMP)-activated protein kinase (AMPK) by physical exercise, release of leptin, adinopectin from adipose tissue or pharmacological means (thiazolidinedione and metformin) improve insulin action through the inhibition of iNOS as well as mTOR/S6K1 signaling. Simultaneously, AMPK can also increase glucose transport by triggering GLUT4 translocation. The protein tyrosine phosphatases (PTPs), protein tyrosine phosphatase 1 B (PTP1B), leukocyte antigen-related (LAR) and Src homology 2 (SH2) domain-containing protein-tyrosine-phosphatase-1 (SHP-1) promote and develop insulin resistance by dephosphorylation of tyrosine residues within the insulin receptor. Thus, activation of the mTOR pathway promotes insulin resistance, whereas inhibitory action on these pathways can improve insulin resistance.
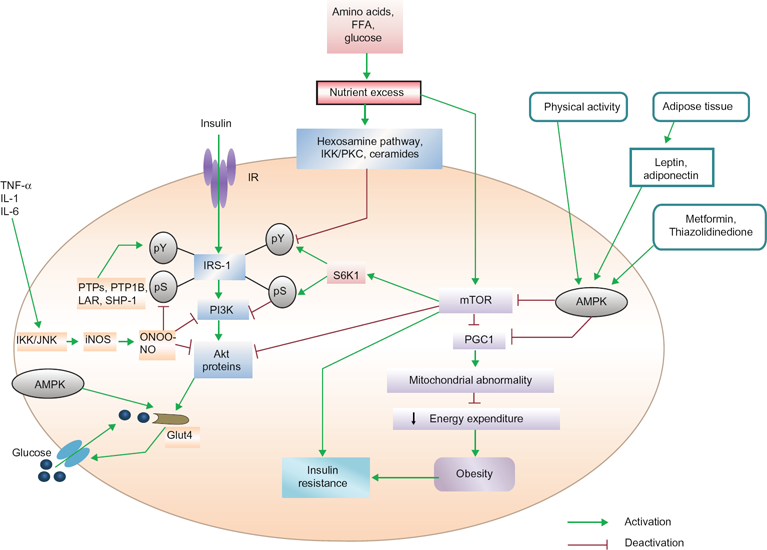
mTOR/S6K1, AMPK and SHP-1 pathways in the development of insulin resistance.
AMPK pathway in insulin resistance
AMPK is a member of a metabolite-sensing protein kinase family, which monitors the energy level of different cells. 42 When energy stored in the cells is decreased by AMPK, the functioning of adenosine triphosphate (ATP)-consuming pathway is stopped and that of alternative pathways for ATP regeneration is started. 43 By activating AMPK, peroxisome proliferator-activated receptor gamma (PPARγ) agonists inhibit iNOS induction in macrophages, myocytes and adipocytes. 44 AMPK switches on the metabolic pathways while turning off inflammation in insulin target tissues and macrophages. The antidiabetic drug thiazolidinedione activates AMPK, which inhibits PPARγ, leading to reduction of PGC1 expression. AMPK is also activated by muscle contraction, exercise and the release of leptin and adinopectin from adipocytes. On the other hand, insulin sensitivity is improved by AMPK because it inhibits the activation of the mTOR/S6K1 pathway (Fig. 2). It is also reported that AMPK is activated by the antidiabetic drug metformin by the inhibition of mTOR/S6K1 in different types of cells. 45 AMPK may be a vital therapeutic target as its activation can reduce both inflammatory processes and nutrient-sensing signals, which may play important role against insulin resistance and for type 2 diabetes.
SHP-1 pathway
It has also been reported that the PTPs, PTP1B and LAR are negative regulators of the insulin receptor kinase in liver and peripheral insulin target tissues. In the signaling pathway, pY is a key to insulin signal transduction. PTPs are prominent candidates to negatively regulate insulin action. 46 The protein tyrosine phosphatase SHP-1 is an inhibitor of signal transduction in the insulin-signaling pathway in the liver and skeletal muscles. 47 Animal models with a deficient SHP-1 protein are glucose tolerant and insulin sensitive for glucose metabolism and these animals show increased insulin-signaling through IRS/PI3K/Akt pathway in the liver and in muscle tissues. 48 Therefore, SHP-1 plays a predominant role in regulation of insulin resistance and type 2 diabetes.
Pathway by which free fatty acids promote insulin resistance
Primarily insulin binds to its receptor, which promotes phosphorylation of IRS, then activates PI3K, which in turn activates protein kinase B (PKB)/Akt, and enhances GLUT4 translocation to the cell surface. FFA metabolites [ceramides, acyl-CoA, diacylglycerol (DAG)] activate serine/threonine kinases [PKC, nuclear factor kB (NFkB),inhibitory kB kinase β (IKKβ)], which inhibit IRS and PKB/Akt, thereby inhibiting insulin signaling and preventing GLUT4 translocation. Elevated levels of FFA thereby inhibit glucose utilization by inhibiting the insulin-signaling pathway and reducing insulin-stimulated muscle glycogen synthesis that also occurs in type 2 diabetes mellitus.
Normally, plasma glucose enters the cell by GLUT4. Then glucose is converted to glucose-6-phosphate in presence of Glucokinase. Glucose-6-phosphate is converted to uridine diphosphate (UDP)-glucose and ultimately glycogen in the presence of glycogen synthase, to remain stored as glycogen. Excess FFA in plasma inhibits the IRS signaling pathway and leads to reduced GLUT4 translocation and reduced glucose uptake. As a result, the plasma glucose level is elevated and type 2 diabetes develops. Increased deposition of intramyocellular lipid metabolites (eg, fatty acyl-CoAs, DAG) produce increased cytoplasmic levels of acyl-CoA. These molecules stimulate many serine/threonine (Ser/Thr) kinases, 22 such as PKC, IKK and cytokines, such as NFkB, TNF-α and IL-6. Proteins of the insulin-signaling pathway are phosphorylated by Ser/Thr kinases. IRS-I is a substrate for PKCs such as protein kinase C μ (PKCμ) or protein kinase C α (PKCα). When IRS-I is phosphorylated on Ser-307, it leads to a decreased IRS-I-associated PI3K activity. As a result, IRS-I inhibition decreases GLUT4 activity and glucose uptake (Fig. 3). Glycogen synthase kinase and PKB/Akt are two important proteins of the insulin-signaling pathway that are inactivated through PKC-dependent mechanisms. 49 In mice, inactivation of protein kinase C ζ (PKCζ) was shown to prevent insulin resistance induced by a high-fat diet. 50 As discussed above, IKK and NFkB are two other main Ser/Thr kinases activated by lipid metabolites and inflammatory molecules. 51 Many studies reported a strong association between oxidative stress and insulin resistance. 52 It is also known that the accumulation of intramyocellular fatty acid metabolites produces free radical reactive oxygen species (ROS). 53 Recent evidence indicates that insulin resistance also occurs in pancreatic β-cells, leading to type 2 diabetes. According to the ‘lipotoxicity’ hypothesis, chronically elevated FFA may directly damage the cells of the pancreas by increase NO production. Evidence from transgenic mice expressing human islet amyloid polypeptide (IAPP) indicates that increased dietary fat consumption has a direct link with the pancreatic β-cell dysfunction by inducing islet amyloid deposition which promotes insulin resistance and ultimately type 2 diabetes is developed.54,55 High plasma FFA and increased intracellular lipid concentration inhibit insulin signaling in muscle. An increased level of FFA has a toxic effect on pancreatic β-cells and develops insulin resistance, by which a secondary pathogenetic mechanism of type 2 diabetes can be elucidated.
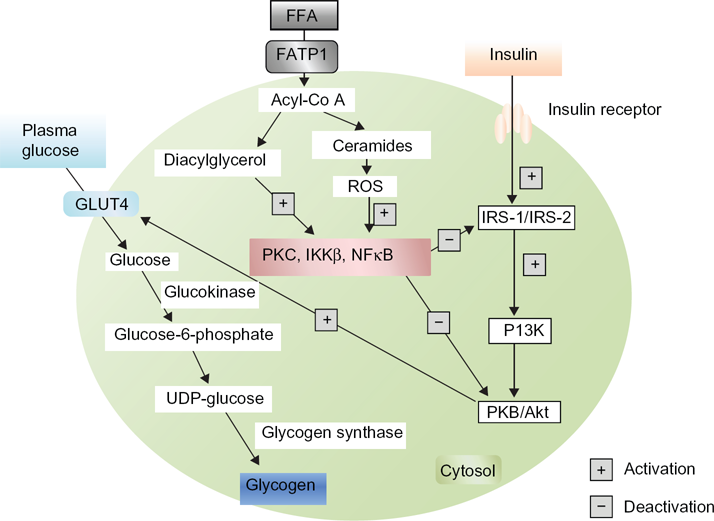
Inhibitory pathway of GLUT4 translocation by free fatty acids.
Differentiation between Insulin Resistance and Type 2 Diabetes
An association between insulin resistance and type 2 diabetes has already been established. 56 Insulin resistance is not only one of the most powerful predictors of the future development of type 2 diabetes, but is also a target of therapeutic approaches once hyperglycemia is developed. 57 Insulin primarily increases the cellular uptake of glucose. During the development of insulin resistance, hepatocytes, adipocytes and myocytes do not respond well to insulin. The β-cells of pancreatic islets increase insulin release sufficiently to overcome the reduced efficiency of insulin action to maintain normal glucose tolerance. Thus, people with insulin resistance also have hyperinsulinemia. 58 When cells are unable to take up glucose well, glucose accumulates in the blood. Insulin resistance thereby causes elevated fasting blood glucose as an indication of type 2 diabetes. Insulin resistance increases the risk of developing type 2 diabetes. Type 2 diabetes mellitus (NIDDM) is a complex metabolic disorder that is characterized by high blood glucose in the context of insulin resistance as well as relative insulin deficiency. Fluctuations in insulin sensitivity occur throughout the normal life cycle including puberty, pregnancy and aging.59–61 Prediabetes usually occurs in people who already have insulin resistance. Normal β-cells produce an adaptive response to insulin resistance, which results in increasing numbers of β-cells in the islets and enhancement of their function to release more insulin, and normal glucose level is maintained. Although insulin resistance alone does not cause type 2 diabetes, it often sets a stage for the disease by placing high demand on the insulin-producing β-cells, which results in a gradual loss of functions of β-cells and the development of type 2 diabetes. Both obesity and type 2 diabetes are associated with insulin resistance. Some obese and insulin-resistant persons do not develop hyperglycaemia, 62 whereas in similar persons with type 2 diabetes, β-cells are unable to release insulin more in response to decreased insulin sensitivity of the cells.63–65 Eventually, β-cell dysfunction occurs, resulting in an impaired glucose tolerance and an enhancement of fasting blood glucose, eventually leading to type 2 diabetes. 66 NEFA are potential molecules to link insulin resistance and β-cell dysfunction and apoptosis (Fig. 4) in individuals with type 2 diabetes. 67 In order to provide energy to cells during insulin resistance, lipolysis occurs, which provides a high plasma level of NEFA. A high plasma level of NEFA causes lipotoxicity and progressive loss of β-cell function. NEFA also decrease normal insulin release from β-cell with a marked impairment in glucose-stimulated insulin secretion in them and reducing insulin biosynthesis. A progressive loss of β-cell function causes impaired glucose tolerance in normal cells, eventually leading to insulin resistance in those cells and then to type 2 diabetes. 69 Thus, development of insulin resistance may be a prediabetic condition playing a pivotal role for the process of development of type 2 diabetes.
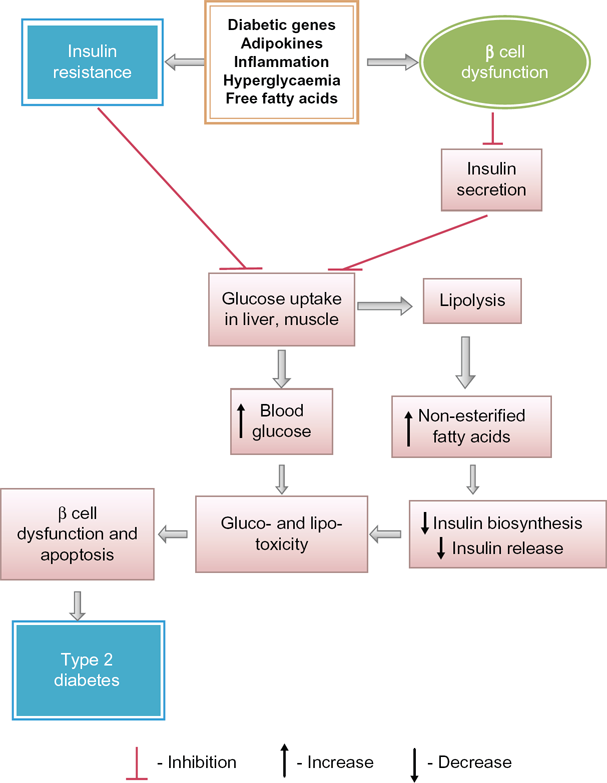
Predominant biochemical alterations for progression of type 2 diabetes from insulin resistance and due to beta cells dysfunction.
Conclusion
An association among elevated FFA, obesity, peripheral insulin resistance and development of type 2 diabetes has been elucidated. In obese individuals, plasma FFA not only produces insulin resistance in skeletal muscles, but also may have additional actions in the liver and the pancreas, which lead to the development of type 2 diabetes. Obesity results in a pro-inflammatory state starting from the metabolic cells and also recruiting immune cells with the release of inflammatory cytokines (TNF-α, IL-6, adiponectin, etc). These inflammatory mediators cause obesity-induced inflammation, the chronic form of which generally leads to complications such as insulin resistance and diabetes mellitus. In type 2 diabetes, excess synthesis of lipid droplets occurs and a formation of lipid peroxides becomes hazardous to health, since it may aggravate the prevalence of a diseased condition or generate new diseases (cardiovascular diseases, cancer, etc) in the body. Thus, prevention of lipid peroxidation is a weapon to slow down the process of development of diabetic complications and other diseases. Ectopic lipids (DAG and possibly ceramides) may be the root cause of liver and muscle insulin resistance and new therapies aimed at decreasing the lipid contents in these organs may represent efficacious therapeutic targets for the treatment of insulin resistance and its associated comorbidities. At a societal level, concerted efforts to restore balance in our diets and behaviors (sedentary life-style, smoking, drinking alcohol, psychological stress, etc) can prevent obesity and ectopic lipid deposition. The involvement of lipid in the development of diabetic complications can no longer be deniable. In the end, lifestyle modifications such as physical activity, diets, change of sedentary to active life format may not be enough to control diabetes, the metabolic disease, in obese patients. Instead, more research is required to find the molecular targets to achieve proper therapeutic strategies to combat type 2 diabetes, the notoriously slow killer.
Footnotes
Author Contributions
Contributed to the writing of the manuscript: BM, CMH, LM, PP, MKG. Agree with manuscript results and conclusions: BM, CMH, LM, PP, MKG. Jointly developed the structure and arguments for the paper: BM, CMH, LM, PP, MKG. Made critical revisions and approved final version: BM. All authors reviewed and approved of the final manuscript.
Funding
We are indebted to Department of Science and Technology (Govt. of India) grant no. DST/inspire/fellowship/2010(87) and Indian Council of Medical Research, Grant no. 58/7/2009-BMS, for this work.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests. Provenance: the authors were invited to submit this paper.