
Abstract
Argyrophylic grain disease (AGD) is a neurodegenerative condition that has been classified among the sporadic tauopathies. Entities in this group present intracellular aggregates of hyperphosphorylated tau, giving rise to characteristic neuronal and glial inclusions. In different tauopathies, the proportion of several tau isoforms present in the aggregates shows specific patterns. AGD has been tentatively classified in the 4R group (predominance of 4R tau isoforms) together with progressive supranuclear palsy and corticobasal degeneration. Pick's disease is included in the 3R group (predominance of 3R isoforms), whereas tau pathology of Alzheimer's disease represents and intermediate group (3 or 4 repeats [3R plus 4R, respectively] isoforms). In this work, we have analyzed tau present in aggregates isolated from brain samples of patients with argyrophylic grain disease. Our results indicate that the main tau isoform present in aggregates obtained from patients with AGD is a hyperphosphorylated isoform containing exons 2 and 10 but lacking exon 3.
Introduction
Argyrophilic grain disease (AGD) is a neurodegenerative condition associated with old age and characterized by the presence of abundant small spindle 4 to 8 μm argyrophilic inclusions (argyrophilic grains) in neurons of the amygdala, entorhinal cortex, hippocampus, and other limbic brain regions. 1 3 The neuropathological profile of AGD has been described in detail, and a highly homogeneous regional distribution of lesions allows a classification of cases according to a staging system. 4 Together with the characteristic argyrophylic grains (AG), all cases display a high density of diffusely taupositive neurons (pretangles) and numerous ballooned neurons. Additionally, oligodendroglial tau inclusions (coiled bodies) and astrocytic inclusions (bush-like astrocytes) are invariably found in involved areas. Since argyrophylic grains and associated neuronal and glial inclusions are immunoreactive with tau antibodies, 5 7 AGD has been classified as a sporadic tauopathy. Tauopathies have been characterized by the presence, mainly in protein aggregates, of tau protein with either 3 or 4 repeats (3R or 4R, respectively) in the tau microtubule binding domain. 8 Tau aggregates from Alzheimer's Disease (AD) are composed of similar amounts of tau 3R and tau 4R isoforms. 9 In contrast, corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP) 10 present an increase in the proportion of tau 4R, whereas Pick's disease (PiD) contains predominantly tau 3R isoforms in tau aggregates. 11 Previously, it has been suggested that AGD could be a tau 4R tauopathy. 2 Western blot analysis of tau isoforms in pure AGD has shown a double band at 64 and 69 kDa and a minor 74 kDa band, suggesting a predominance of 4R tau isoforms. Accordingly, AGD together with CBD and PSP may be considered collectively as 4R tauopathies. In fact, cases with mixed phenotypic features (eg, PSP plus CBD, or AGD plus PSP or CBD) are not infrequently observed.
AGD seems to be a sporadic disease. However, the presence of AGD pathology has been described in a patient bearing a mutation in tau gene, MAPT S305I. 12 Genetic risk factors for AGD show clear differences from other neurodegenerative dementias like Alzheimer's disease. A higher frequency of apolipoprotein E isoform 2 has been reported in AGD patients,13,14 although this point has been questioned.15,16 Additionally, it has been discussed whether the presence of MAPT haplotype H1 could be significantly increased in AGD. 17 As for other neurodegenerative dementias, aging appears to be the main risk for AGD, which is particularly prevalent in the oldest old. It has been estimated that one-third of centenarians may present AGD. 18
Given that the presence of hyperphosphorylated tau is a dominant feature in the neuropathological and molecular profile of all tauopathies and that most probably it plays a central role in pathogenesis, some clues to the specific phenotype of AGD may be related to its putative molecular signature. However, the interpretation of molecular studies in AGD cases has been hampered by several limitations: (1) most often AGD is found in combination with other neurodegenerative pathologies and is particularly associated with other 4R tauopathies; (2) AGD involves predominantly the medial temporal lobe, a brain region where pathology of the Alzheimer's type is highly prevalent in old subjects; and (3) pure AGD cases are rare and show atypical clinical features, such as rapid progression, and postmortem studies are frequently performed under a clinical suspicion of Creutzfeldt-Jakob disease (CJD). Availability of fresh frozen tissue for molecular studies is usually limited when high-risk neuropathological autopsies are performed.
Now, in this work we have analyzed the composition of the main tau isoform present in tau aggregates obtained from a series of well-characterized cases of AGD presenting either alone or combined with other neurodegenerative conditions.
Materials and Methods
Cases
Main characteristics of the patients analyzed in this study.

Rapidly progressive dementia, studied postmortem as suspected CJD.
Tau haplotypes (H1 and H2) and APOE isoforms (E3 or E4) are indicated.
Neuropathology
In all cases, postmortem examination was limited to the cranial cavity (with spinal cord extraction only in cases with ALS) with a postmortem interval shorter than 12 hours. According to the brain bank protocol (shared by both brain banks involved), in conventional donation cases, immediately after extraction, the right half of the brain was sliced and frozen, while the left half was fixed by immersion in phosphate- buffered 4% formaldehyde for at least 3 weeks. High-risk autopsies for suspected CJD were performed following international recommendations. In these cases, fresh tissue blocks from the right frontal and parietal lobes and the ipsilateral cerebellar hemisphere were obtained for freezing. All frozen tissue was stored at -80°C. After fixation, a full neuropathological study was performed in the left half brain. For that purpose, 25 tissue blocks were obtained from cortical and subcortical brain regions for parafin embedding. Neuropathological classification of cases was based on a thorough examination of hematoxylin-eosin stained paraffin sections of all blocks, and immunostaining with a panel of antibodies (beta-amyloid, tau AT100, alphasynuclein, ubiquitin, prion protein, and TDP-43) in selected sections. Consensus criteria were used for disease diagnosis and staging.
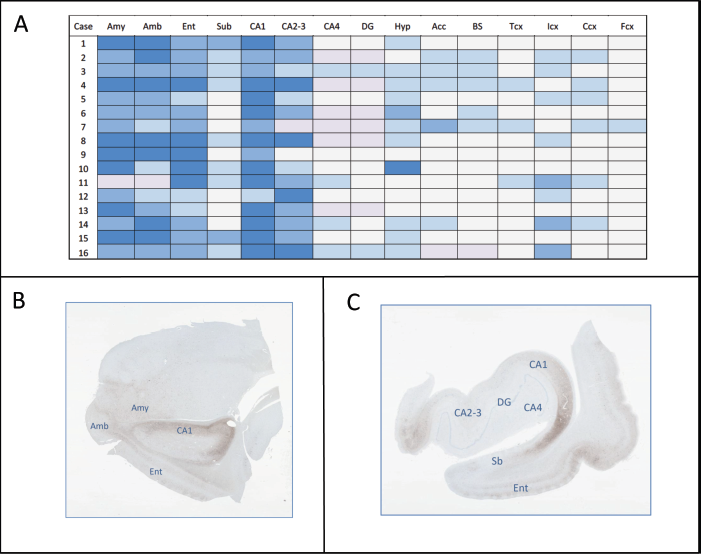
Further morphological study of AG pathology was performed in 2 tissue blocks comprising (1) amygdala and (2) anterior hippocampus. Paraffin sections of these blocks were studied with Gallyas stain and with immunostaining for a panel of antibodies (tau AT8, tau AT100, tau 7.51, RD3, RD4, neurofilaments SMI 1, p62). The presence and intensity of various tau-reactive inclusions in several regions was assessed on a semiquantitative basis on AT8-immunostained sections (Fig. 3A). For that purpose, the frequency of AG was graded on a 0 to 3 scale. Other tau-immunoreactive inclusions associated to AG, principally pretangles and coiled bodies, show parallel variations in frequency (data not shown). For the evaluation of ballooned neurons anti-tau antibodies and antibodies recognizing hyperphosphorylated neurofilaments (SMI 1) (1:100) (Sternberger Monoclonals, Covance Inc., Princeton, New Jersey) were employed.
Antibodies
Tau antibodies used in this study. Localization of the antibodies that react with (
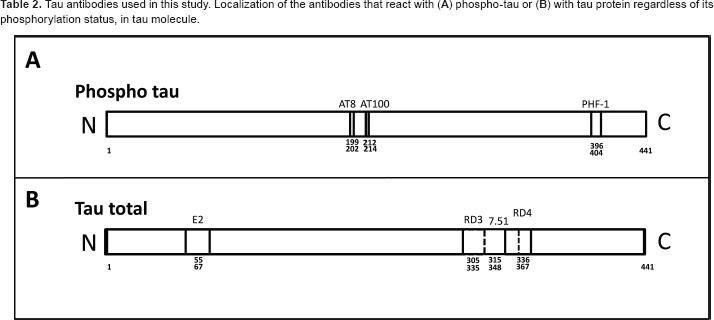
PHF-1 (1:100), an antibody that reacts with tau when Ser396 and Ser404 are phosphorylated, was a kind gift of Peter Davies, Ph.D. (Albert Einstein University, NY, USA). Exon-2 (1:100) antibody was raised against human tau peptide comprising the residues 55 to 67 (numbering of the longest human tau isoform, 20 and it was obtained through Abyntek Biopharma S.L., (Derio, Spain) for our laboratory (R. Cuadros et al, unpublished data, 2011). Finally, anti-tau clone 8E6/C11 (1:500) and anti-tau clone 1E1/A6 (1:500), specific for reacting with tau 3R and tau 4R isoforms were from Merck Millipore, Billerica, Massachusetts.
Immunohistochemistry
Immunohistochemical stains were performed on 5 μ paraffin sections by using the ABC method (Pierce Antibodies, Thermo Fisher Scientific Inc., Waltham, Massachusetts), diaminobenzidine (DAB) as developer, and Carazzi's hematoxylin as counterstain. Optimal dilutions and times and conditions of incubation of primary antibodies were applied as recommended by the suppliers. Unmasking of antigens was performed when necessary.
Preparation of brain tissue extracts
Brain cell extracts were isolated by homogenizing the brain tissue in ice-cold buffer (1:10 w/v) consisting of 10 mM Tris pH7.4, 1 mM EGTA, 0.1 M NaCl and 10% sucrose plus phosphatase inhibitors (10 mM NaF, 1 mM sodium orthovanadate), and protease inhibitors (2 mM PMSF, 10 μg/mL aprotinin, 10 μg/mL leupeptin, and 10 μg/mL pepstatin).
Isolation of detergent insoluble tau aggregates
Tau aggregates from brain tissue samples of the patients were isolated as previously described for PHF-tau from Alzheimer disease patients by Greenberg and Davies.19,21 Different sources were used to look for tau aggregates: temporal (including medial temporal lobe), frontal, and parietal cortex.
Detergent-insoluble tau aggregates were visualized by electron microscopy. 22
Western blot analysis
Extracts for Western blot analysis were prepared by homogenizing the isolated tau aggregates samples. The samples were homogenized at 4°C and protein content determined by the Bradford method (Bio-Rad laboratories Inc., Hercules, California). Total protein (10 μg) was electrophoresed on 8% SDS-PAGE gel and transferred to a nitrocellulose membrane (Schleicher and Schuell). The experiments were performed using the following primary antibodies: AT8, 7.51, PHF-1, AT100, Tau 3R, and Tau 4R. The filters were incubated with the antibody at 4°C overnight in 5% nonfat dried milk. A secondary HRP-conjugated goat antimouse (Gibco, Life Technologies Corp., Carlsbad, California) followed by ECL detection reagents (Amersham, GE Healthcare Bio-Sciences, Piscataway, New Jersey) were used for immunodetection.
Transmission electron microscopy
Immunoelectron microscopy was performed after adsorption of the samples to electron microscopy carbon-coated grids and incubation with the first antibody (AT8 or PHF-1) for 1 hour at room temperature. After extensive washing, the grids were incubated with the secondary antibody (1/40) conjugated with 10 nm diameter gold particles. Finally, the samples were stained with 2% uranyl acetate for 1 minute. Transmission electron microscopy was performed in a JEOL model 1200EX electron microscope operated at 100 kV. Electron micrographs were obtained at a magnification of x 50,000.
Apolipoprotein E (APOE) and H/H genotyping
Total DNA was isolated from peripheral blood or cerebral tissue following standard procedures. Genotyping of MAPT rs1052553 polymorphism, which discriminates between MAPT H1 and H2 haplotypes, was determined using TaqMan probes (C—7563736_10 assay, Applied Biosystems, Life Technologies Corp., Carlsbad, California) according to manufacturer instructions. APOE genotyping (rs429358 and rs7412) was performed by real-time polymerase chain reaction (PCR). 23
Results
Grains are stained by an antibody that recognizes phospho-tau
Histopathological analysis (Fig. 1A) indicates the presence of characteristic silver stained grains in the brain of the patients. When immunofluorescence analyses were carried out using the AT8 antibody, which recognizes phospho-tau, a clear reaction with the grains was observed (Fig. 1B). Furthermore, immunoelectron microscopy analyses indicate the presence of phospho-tau in grains and filamentous tau aggregates (Fig. 1C). These filamentous polymers were also stained when another antibody, reacting with a different tau phosphoepitope (PHF- 1), was used. Figure 1D shows the filaments in the absence (left and middle) or presence (right) of anti-PHF-1 staining (Fig. 1D). Nevertheless, the proportion of AGD filamentous polymers found in a brain region such as the temporal lobe is much lower than that found in the same region in other tauopathies such as AD (Fig. 2).
AGD aggregates. Fibrillar polymers from a cell extract of temporal lobe of (

Brain regions containing argyrophylic grains
In all cases and all regions examined, AG were highly associated with other histological lesions characteristic of AG pathology: ballooned neurons, diffusely tau-reactive neurons (pretangles), neurofibrillary tangles, coiled bodies, and tau-reactive astrocytes, frequently showing bush-like morphology. A consistent pattern of regional distribution of AG pathology was identified both in cases with a principal diagnosis of AGD (patients 1 to 6) and in cases with AGD combined to different neurodegenerative conditions, including other sporadic tauopathies (patients 7 to 16). Areas most intensely involved in the medial temporal lobe were the amygdaloid complex (predominantly the basolateral nuclei), gyrus ambiens, entorhinal and perirhinal cortex, and CA1 sector of the hippocampal cortex. In contrast, CA4 sector and the dentate gyrus were proportionally less affected by AG pathology. Most cases (14/16) were classified as stage III AGD, and cases with a main diagnosis of AGD displayed more intense pathology in the hypothalamus, nucleus accumbens and brainstem tegmentum (Fig. 3A).
Characterization of the main tau isoform present in AGD tau aggregates
Six tau isoforms are present in the human central nervous system (CNS) (Fig. 4A) (see Goedert
24
and Goedert et al
25
). Characterization of tau isoforms present in aggregates, such as the paired helical filaments found in Alzheimer disease, reveals, by electrophoresis and Western blot analysis, the presence of 3 main bands such that each corresponds to two different phospho-tau isoforms (Fig. 4B).

When tau isoforms present in AGD aggregates were analyzed by electrophoresis/Western blot, mainly a band with a relative electrophoretic mobility corresponding to a protein of 64K was observed. Figure 4C shows the electrophoretic patterns of tau aggregates from different AGD patients using 7.51 antibody. In all cases, the 64K band was the most abundant band, which could result from the addition of the 2 tau isoforms containing exon 2, one of them containing and the other one lacking exon 10 (Fig. 4D). When Western blot was carried out to check for the presence or absence of exon 10, using specific antibodies that recognize tau 3R or tau 4R isoform, only tau 4R isoform was found in the 64K band for AGD aggregates. In addition, this band also reacts with an antibody raised against residues 55 to 67 (nomenclature of the largest tau isoform present in the CNS) located at exon 2 (Fig. 4D). Tis pattern was consistently found in patients with pure AGD and in cases with AG pathology combined with other neurodegenerative conditions, including 4R tauopathies. Tus, we conclude that the major tau isoform present in AGD aggregates is that containing exon 2 and exon 10 and lacking exon 3.
Status of APOE and H/H polymorphisms
Genotyping for APOE and MAPT H/H haplotypes was performed in 12/16 cases (Table 1). All patients in this study were APOE e3/e3 (10/12, 83,3%) or APOE e3/e4 (2/12, 16,7%). No e2 cases were observed. These findings are in contrast with previous reported series based in central European populations where a significantly higher proportion of AGD cases contained the e2 haplotype.13,14,16 Noteworthy, a large series based in a North American population did not confirm this association. 15 Although the present series includes a proportionally shorter number of cases, the predominance of the e3 haplotype, with a high proportion of homozygosis, may depend on the higher prevalence of this haplotype in southern European populations. 26 In the present series, 9 out of 12 cases (75%) contained the H1/H1 haplotype at the MAPT gene, while 3/12 cases were H1/H2. These results agree with previous studies, where a high proportion of H1/H1 haplotype has been described in association with sporadic 4R tauopathies, including AGD. 17
In general, it is not clear how APOE isoforms or MAPT haplotypes might condition the development of a tauopathy like AGD. Clinical subtypes of other tauopathies like frontotemporal lobar degeneration (FTLD-Tau), display different tau and APOE genotype frequencies. 27
On the other hand, APOE isoforms may regulate tau kinases and phospho-tau levels in neurons. 28 Additionaly, MAPT H1 haplotype is more efficient at driving gene expression than the H2 haplotype, and this suggests that when H2 haplotype is present a lower amount of tau protein is expressed. 29 Tus, differences in APOE isoforms and MAPT haplotypes may regulate the amounts of phospho-tau and total tau, two important parameters in the onset of tauopathies. How both features could affect the development of AGD remains under discussion, as previously indicated.
Discussion
AGD may manifest with dementia and behavioral abnormalities such as emotional and mood imbalance.3,30 These behavior disorders may correlate with the appearance of tau pathology (silver stained or argyrophylic grains) at hippocampal formation and amygdala regions. On the other hand, temporal- spatial spreading of tau aggregates has been described in AGD. 4 In this work, we have found a regular pattern of involvement of anterior medial temporal lobe regions by AG pathology with further extension to limbic cortices, certain subcortical nuclei, and brainstem tegmentum. However, in our series, AG pathology usually spares the dentate gyrus, a region that could be rich in tau 3R isoforms due to the presence of adult neurogenesis. Regional distribution of pathology was found to be comparable in pure AGD cases and in AGD pathology associated with other neurodegenerative conditions but with a higher intensity of involvement of extratemporal regions in pure cases. We also found that tau aggregates obtained from patients with AGD are composed predominantly by a tau isoform containing exon 2 and exon 10 and lacking exon 3. Tis finding was consistent in patients (1) with pure AGD, (2) with AGD associated with another 4R tauopathy, and (3) with AGD combined with other neurodegenerative diseases with low Braak stages for Alzheimer's type neurofibrillar degeneration. Previous studies 31 33 have reported a double-band of tau at 64K and 69K in AGD, thus including AGD in the 4R tauopathies group together with PSP and CBD. Our findings suggest that AG pathology may be characterized by a specific pattern of tau isoforms expressed in Western blot by a predominant band at 64K.
Recently, it has been shown that the exon 2 and exon 10 inserts increase tau aggregation propensity, whereas the exon 3 insert decreases tau aggregation. 34 Also, the presence of H2 haplotype together with the expression of exon 3 seems to play a protective effect against the development of some tauopathies.35,36 However, in our work we found that one-fourth of our AGD patients show the H2 haplotype. Although the number of cases studied in this series is too low to draw strong conclusions, our data indicate a lower proportion of H2 haplotype compared with normal controls in a similar population. 37
In summary, we have shown that tau aggregates of AGD enclose as a major component the phosphorylated tau isoform containing exon 2 and 10 and lacking exon 3. Phosphorylation of tau occurs, at least, in those sites recognized by antibodies AT8, AT100, and PHF-1. Despite this posttranslational modification, other modifications have been described for tau protein, and recently, one of these modifications, acetylation, appears to be absent in AGD, 38 whereas it is present in other tauopathies like Alzheimer's disease.
Author Contributions
Conceived and designed the experiments: AR, FH, JA. Analyzed the data: AR, MC, JA. Wrote the first draft of the manuscript: AR, RC, JA. Contributed to the writing of the manuscript: MC, FH. Agree with manuscript results and conclusions: AR, RC, MC, FH, JA. Jointly developed the structure and arguments for the paper: AR, JA. Made critical revisions and approved final version: AR, FH, JA. All authors reviewed and approved of the final manuscript.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.