
Abstract
Vanishing white matter (VWM) leukoencephalopathy is one of the most prevalent hereditary white matter diseases. It has been associated with mutations in genes encoding eukaryotic translation initiation factor (
Introduction
Vanishing white matter disease (VWMD), also known as childhood ataxia with central hypomyelination (CACH), 1 is one of the most prevalent hereditary white matter diseases. 2 It typically appears in a previously healthy child, and it produces a progressive neurological deterioration with some acute exacerbations in reaction to certain stimuli, such as infections, cranial trauma, and acute fright.3,4 It can also produce optic atrophy and progressive macrocephaly. 5
The most frequent clinical forms of the disease have their onset during childhood: the early childhood-onset appears before five years of age and the late childhood-onset between ages 5 and 15.4,6 The disease may have a chronic progressive course or an acute form, and it is difficult to predict the evolution that each patient will follow even though some efforts to find a correlation between genotype and phenotype have been made. As published before, the evolution can be different even among affected members of the same family. 6
MRI images show important diffuse abnormalities of the signal on T1 and T2 of almost all the cerebral white matter, with progressive rarefaction and cystic degeneration that leads to its complete disappearance. These findings are better seen on proton density and Fluid attenuated inversion recovery (FLAIR) images, where within the rarified and cystic white matter a stripe-like pattern can be found, which represents remaining tissue strands.
Proton magnetic-resonance spectroscopy shows normal signals at the beginning, but they progressively disappear and a spectrum similar to that of cerebrospinal fluid (CSF) can be recognized.7–11 Sparing of the U fibers can be found to a variable extent, and it is best seen on T1-weighted images.
It is remarkable that even in the end-stage, when almost all white matter is cystic, atrophy is usually mild if any.
These abnormalities of the white matter are unique and completely different from those seen in other neuropsychiatric disorders because they diffuse and are homogeneous affecting brain and cerebellum. 12
Radiological criteria for the diagnosis of VWM have been described for MRI studies such as Van der Knaap et al, 4 and there is an MRI-based approach to the diagnosis of leukodystrophies made by Schiffmann and Van der Knaap in 2009. 13
Other examinations are of little help in the diagnosis of VWM. There has been reported to be only a slight increase in glycine level in CSF, with a glycine CSF/plasma ratio mildly elevated, much lower than that of the nonketotic hyperglycinemia, but this determination has not proved either to be constant through all the evolution of the disease or present in all confirmed patients.6,14
EEG is normal at the beginning of the disease and progressively shows diffuse lentification of basal activity with multifocal paroxysms. Visual- and auditory-evoked response potentials appear normal at onset and later show an increase in latencies until disappearance of all waves. All these findings are unspecific and can be seen in several neurodegenerative diseases. Electromyography and motor conduction velocity are normal, in contrast to other leucodystrophies such as metachromatic leukodystrophy. 6
Genetic studies have proved an association between this disease and molecular mutations in five genes (
It is known that
Histopathological studies confirmed the diagnosis, though nowadays, thanks to advances in genetics and neuroimaging, it is not necessary to take cerebral biopsies or necropsy studies to diagnose this disease.
Until now no curative treatment has been found. What seems to be effective is the prevention of the acute deterioration, avoiding cranial trauma and treating infection aggressively because this seems to prevent or at least foreshorten the deterioration associated with the disease, and it may increase survival.1,6 Nevertheless, chronic progression is unavoidable.
Molecular genetic studies are of great help in confirming the diagnosis and providing genetic counseling and prenatal diagnosis to the families.
Our aim was to register all the cases diagnosed to date in Spain and compare them to other series.
Material and Methods
We have retrospectively studied all cases with clinical findings, neuroimaging and, more recently, genetics that appear to be compatible with VWMD in the last 30 years. All information was confidentially compiled according to the Declaration of Helsinki, and no personal data were used. We found 21 patients throughout the country; 2 of them were diagnosed 30 years ago after a necropsy study. We obtained and re-examined some samples from their necropsies. The next 3 were diagnosed by both clinical and radiological criteria, following van der Knaap radiological criteria, 4 and in recent years, 16 were genetically confirmed. We revised clinical signs, complementary study outcomes, and survival of all 21 children.
The first patients (cases 1, 2, 3, and 4) were diagnosed before 1980s, and they underwent tomographies and necropsies. The rest of the children have magnetic resonance images although some of them have tomography images too. When collecting the images, we chose T1 and T2-weighted MR images to show abnormalities in the intensity of the white matter, and FLAIR images to show the cystic degeneration in it. Besides, we collected information of spectroscopy studies when available.
Regarding genetic studies, they were mostly carried out in the Department of Pediatrics and Child Neurology, VU University Medical Center, Amsterdam, Netherlands Van der Knaap et al, and consisted of sequence analysis of the entire coding region of the five genes. Once in a child was found a mutation, genetic family testing was performed. Sequencing of primers is described elsewhere. 20
Finally, with respect to necropsy studies, we found it difficult because only few samples from patients 1 and 2 were available and dated from 1980s. We examined macroscopic and microscopic samples, being the latter preserved in two kinds of staining: hematoxylin—eosin staining and Kluver—Barrera staining.
We are aware that we were limited by the fact that some cases were retrospectively compiled, and we only had medical reports in some of them.
Results
We found 21 confirmed VWMD cases, 13 girls and 8 boys. Age at diagnosis was between 18 months and 8 years. Among all these patients, there were 3 children (2 girls and 1 boy) who could be cataloged in the late-childhood-onset form (with 5.5, 6, and 8 years of age at the first clinical signs, respectively). The rest were included in the early childhood-onset form. No prenatal or juvenile onset forms have been found in our country to date.
There were two pairs of affected siblings; one of these pairs had consanguineous parents. The rest of the children did not have affected brothers or sisters but one of them had a brother who died at a premature age of unknown cause.
None of these children had antecedents of personal history but in three a mild developmental delay (motor delay in two and speech delay in one of them) had been observed.
Among them, 10 children have survived to the present date, with ages from 2.7 to 13 years. A total of 10 children died at 2.1–32 years of age. The longest survival was that for a patient who was diagnosed with VWMD at 2 years of age and died at 32 years of age.
Most of our patients (16 out of 21) suffered a chronic progressive course with episodes of deterioration, and 5 presented an acute and rapid course. Children with chronic progressive course underwent from 1 to 12 episodes of deterioration (mean 6.6 episodes). These episodes were because of banal infections (17 patients suffered at least one) or mild cranial trauma (15 children). Only one child was reported to have episodes secondary to intense fright. In all, 7 of the 15 children with a chronic course had already died after 4–30 years of progressive deterioration.
Among the patients who followed an acute course, four have already died, all of them during the course of a banal respiratory infection, which led in two cases to a status epilepticus with a quick deterioration and death a few days after. The fifth one is still alive but it is only seven months on from his diagnosis.
The most important clinical findings were progressive pyramidal signs. At the onset of the disease, 16 of the 21 patients showed progressive ataxia (6 of them considered to be “severe”), one girl had bilateral palpebral ptosis, another girl had dystonia, and yet another one showed hemiparesis, but the main symptom in all of them was progressive spasticity.
In all, 16 patients lost autonomous deambulation between 18 months and 11 years of age. Five children are still able to walk (2–12 years of age at present).
Cognitive functions were preserved some time after motor loss was established. Children with a rapid progression of the disease began the cognitive impairment when they were 2–4 years old, while patients with a chronic course had normal functions until 6.5–16 years of age. We have nine children still with normal cognitive function, ranging in age from 2.5–12 years.
Nine patients suffered seizures during the evolution of the disease. Three of them had a rapid progression, and they had seizures at 2–3 years of age; the other six patients followed a more chronic course with epilepsy at 4–12 years of age. Mainly they were generalized tonic—clonic seizures, which may have had a focal origin in some patients, and the EEG showed from normal to very slow activity with paroxystic events. There were two children who developed a status epilepticus in the course of a banal respiratory infection resistant to antiepileptic treatment.
Other clinical signs found were optic atrophy and macrocephaly. Optic atrophy was detected in four children, unrelated to age at onset or type of evolution. We also detected progressive macrocephaly in three children.
All patients underwent neuroimaging. The first four cases, who were diagnosed in the 1970s and 1980s, underwent cranial tomography. The rest of the children underwent one or more brain MRI (15/21) (Figs. 1 and 2). These examinations showed an important, diffuse alteration of the white matter signal, with cystic degeneration in children at the end-stages.

(

(
Proton magnetic-resonance spectroscopy showed stagedependent abnormalities; in the initial stages, the white matter signals appeared normal, but at the final stages signals of normal white matter had disappeared and only a spectrum similar to that of the CSF is seen in chronic forms.
In the 1980s, glycine determinations in CSF and blood in four patients and a glycine CSF/plasma ratio were mildly elevated in all of them (0.055–0.092).
Genetic studies showed mutations in the
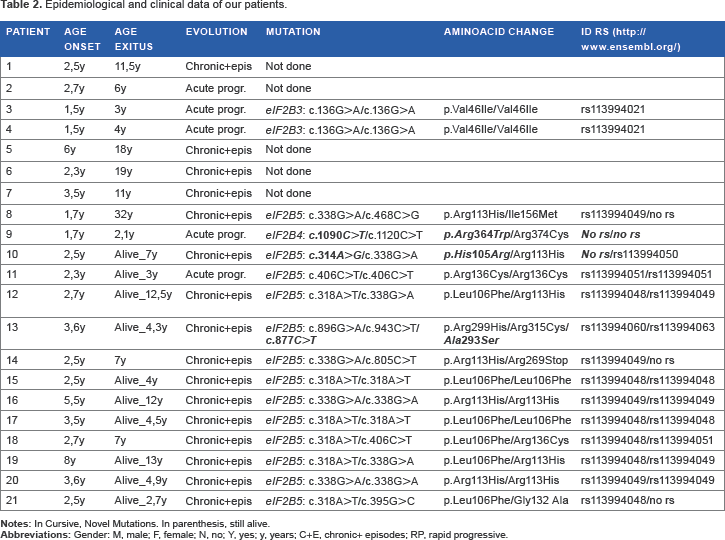
Summarizes clinical and ancillary data of our patients.

We also detected two children with mutations in
We also have found novel mutations: c.1090C>T in
Epidemiological and clinical data of our patients.
No genetics studies could be made on five patients who died before 2001 as DNA was not available. The diagnosis of the first two children was made with necropsy study and in the latter three cases with the radiological and clinical criteria.
The necropsy findings were consistent with previously reported and included brain atrophy, which spared both the cortex and the cerebellum and was associated with ventricle dilation, with a decrease in the white matter of the centrum semiovale and the corpus callosum, while some of the U fibers were better preserved. Microscopically, we found that the cortex was quite preserved, but the affected white matter showed important myelin loss, vacuolation, and cystic changes, with some glial septa inside the cysts. There was also an infiltration of T lymphocytes and astrocytes, which mostly affected the white matter and also the cortex. The cerebellum was less affected (Figs. 3–6).

Patient n.1: macroscopic detail: hemispheric white matter with cystic degeneration from subcortical to periventricular areas.

Patient n.1: microscopic detail: this image shows a well-preserved cortex (circle), with underlying white matter destruction. In the rarefacted white matter, some glial septum appears (small arrows). There is also a disappearance of a part of U-fiber (big arrow).

Patient n.1: hematoxylin—eosin staining: cellular infiltration inside the glial septum; some glial septa are marked with arrows. These cells are shown as small, dark, round nucleus and appear to be oligodendrocytes.

Patient n.1: Kluver–Barrera staining: glial septum inside the white matter cysts. Some glial septa are marked with arrows. This staining highlights myelin fibers.
We tried corticosteroids in the first three patients to diminish the potential inflammation secondary to the white matter destruction, but we saw no clinical improvement. We did not try this treatment in the rest of the patients because of its limited benefits and adverse effects.
Discussion
VWM leukoencephalopathy is a disease of cerebral white matter. It was described by Van der Knaap and colleagues in 1996 after the finding of cystic degeneration of the cerebral white matter in the necropsy of nine patients. Some case reports in the 1960s and 1970s might correspond to this disease.1,7,21,22
VWM disease has an autosomal recessive mode of inheritance. In our series, we found two pairs of siblings, and the family of one of these pairs was consanguineous. Nevertheless, some autosomal dominant forms have been reported, such as one described by Labauge et al in 2005. 23
Typical signs and symptoms comprise an initial motor deterioration, with pyramidal signs and cerebellar ataxia, with the pyramidal signs being the most evident. Cognitive impairment is less evident and appears later. 1 We reported nine patients with normal cognitive functions nowadays, and it may be because they are following a chronic course and only few years have passed since their diagnoses. There can also be epileptic seizures all along the course of the disease; in our cases, 42% of the children suffered from them. Other clinical forms can develop tremor, dystonia, or chorea. Additional signs have been reported, such as optic atrophy (only seen in four of our children) and progressive macrocephaly (seen in three cases in our series). We did not find an association between these findings and the course of the disease, because they appear both in chronic and acute forms with similar proportions. One of our children had bilateral palpebral ptosis at the onset of the disease, which is an infrequent but already described symptom 24 ; another girl showed hemiparesis as her first symptom, which is also uncommon. 25
It still remains to be revealed why clinical signs start at a particular moment though there is evidence of altered white matter signal in magnetic resonance in pre-symptomatic patients 17 ; all our children had suffered from respiratory infections or mild trauma before the onset of the disease, and they had not deteriorated after those episodes. At a particular stage, there is some change that makes the child vulnerable to these episodes. It is still unknown why the course can be very fast or slower, with no clear relation to genetic mutations or age at onset. In our series, as mentioned above, we had a pair of brothers, one of whom followed an acute progressive course and the other a chronic progression.
It is of interest that two of our children underwent a status epilepticus during an intercurrent infection with a rapid deterioration and demise in a few days time. None of the rest of the children suffered from a status epilepticus, but they had shorter seizures after which they partially or completely recovered their cognitive functions. A girl who was diagnosed at 2.5 years of age has to date experienced an acute course and has an important cognitive deterioration with seizures, but she is still alive at 12 years.
Concerning neuroimaging, there is a homogeneous abnormal signal in the cerebral white matter which is already obvious at the onset of the disease (T2-weighted and FLAIR hyperintense signal, and T1-weighted hypointense signal) and remains constant all through the course of the disease, though there can be progressively appearing signs of cystic degeneration. 10
The evolution of the proton MR spectroscopy is typical, showing a spectrum similar to that of the CSF in final stages.9,10,13 We have only observed these findings in the children with a chronic course, perhaps because the more acute forms may lead to death before these changes become evident.
Other examinations are of little help in the diagnosis of VWM. We determined glycine CSF/plasma ratio in four patients, and it was mildly elevated in all of them. We did not repeat this procedure in the rest of the children because this alteration has not been shown to be a consistent marker of the disease, as it has proved neither to be constant through all the evolution of the disease nor present in all confirmed patients.
There have been important advances in the diagnosis of VWM because of genetics since 1996. VWM leukoencephalopathy is caused by mutations in the five genes (
Epidemiological investigations have proved that the most prevalent mutations affect the
We had 13 patients with mutations in
We want to remark upon the relatively high frequency of the mutation c.318A>T corresponding to the amino acid substitution L106H; this is a previously known mutation affecting
In our series, perhaps because it is located in a different country than previous series, it is the second most frequent mutation detected, after the mutation codifying R113H. These children have been diagnosed in different hospitals in several cities, and there is no familial association among them. They come from widely separated cities such as Jaén, Almería and Cordova (Andalusia), Burgos (Castile), and Barakaldo (the Basque Country).
There is evidence of a genotype-phenotype relation, as suggested by previous authors, especially in mutations in
We also found that patients with the mutation c.318A>T (amino acid substitution L106F) all followed a chronic slow course, both in homozygous and heterozygous states. There are no other reports that confirm this observation to date.
No relation has been reported between the
We also found some mutations not described in previous reports: c.1090C>T in
Conclusions
VWM leukoencephalopathy is one of the most prevalent hereditary diseases of white matter in childhood. Clinical and neuroimaging findings are very typical, and they suggest this diagnosis. Molecular genetic studies confirm up to 90% of affected patients. Nevertheless, it is still unknown why this disease starts up at a particular moment and why it follows an acute or chronic form. To date, no curative treatment has been found.
With this report, we want to describe the most frequent clinical and genetic traits of the children diagnosed in Spain and especially highlight the correlation between clinical signs and mutations c.318A>T in
We have described mutations not previously reported: c.1090C>T in
Author Contributions
Conceived and designed the experiments: ETV and MP. Analyzed the data: ETV, MP, VC, ELL, RLP, LGGS, DCM, CSC, NOH, MMG, JAR, VGA, MOC, JM and JAM. Wrote the first draft of the manuscript: ET and MP. Contributed to the writing of the manuscript: ETV, MP, VC, ELL, RLP, LGGS, DCM, CSC, NOH, MMG, JAR, VGA, MOC, JM and JAM. Agree with manuscript results and conclusions: ETV, MP, VC, ELL, RLP, LGGS, DCM, CSC, NOH, MMG, JAR, VGA, MOC, JM and JAM. Jointly developed the structure and arguments for the paper: ETV, MP, VC, ELL, RLP, LGGS, DCM, CSC, NOH, MMG, JAR, VGA, MOC, JM and JAM. Made critical revisions and approved final version: ETV, MP, VC, JM and JAM. All authors reviewed and approved of the final manuscript.