
Abstract
α-Herpesvirinae subfamily members, including herpes simplex virus type 1 (HSV-1) and bovine herpes virus 1 (BHV-1), initiate infection in mucosal surfaces. BHV-1 and HSV-1 enter sensory neurons by cell-cell spread where a burst of viral gene expression occurs. When compared to non-neuronal cells, viral gene expression is quickly extinguished in sensory neurons resulting in neuronal survival and latency. The HSV-1 latency associated transcript (LAT), which is abundantly expressed in latently infected neurons, inhibits apoptosis, viral transcription, and productive infection, and directly or indirectly enhances reactivation from latency in small animal models. Three anti-apoptosis genes can be substituted for LAT, which will restore wild type levels of reactivation from latency to a LAT null mutant virus. Two small non-coding RNAs encoded by LAT possess anti-apoptosis functions in transfected cells. The BHV-1 latency related RNA (LR-RNA), like LAT, is abundantly expressed during latency. The LR-RNA encodes a protein (ORF2) and two microRNAs that are expressed in certain latently infected neurons. Wild-type expression of LR gene products is required for stress-induced reactivation from latency in cattle. ORF2 has anti-apoptosis functions and interacts with certain cellular transcription factors that stimulate viral transcription and productive infection. ORF2 is predicted to promote survival of infected neurons by inhibiting apoptosis and sequestering cellular transcription factors which stimulate productive infection. In addition, the LR encoded microRNAs inhibit viral transcription and apoptosis. In summary, the ability of BHV-1 and HSV-1 to interfere with apoptosis and productive infection in sensory neurons is crucial for the life-long latency-reactivation cycle in their respective hosts.
α-Herpesvirinae Subfamily Members are Important Pathogens in their Respective Hosts
Herpes simplex virus type 1 (HSV-1) and bovine herpes virus 1 (BHV-1) are both important pathogens in their respective natural hosts and both are α-herpesvirinae subfamily members. For example, HSV-1 is the cause of one of the most frequent and serious viral eye infections in the United States, with over 400,000 affected individuals. 1 Following primary infection of the eye, latency is established in sensory neurons within trigeminal ganglia (TG).2,3 HSV-1 reactivates sporadically from TG and the infectious virus can be detected on surfaces of the eye, where it can cause recurrent ocular disease. Reactivation from latency is necessary for recurrent ocular HSV-1 infections.4,5 Long-term oral acyclovir treatment only reduces ocular HSV-1 recurrences by 41%. 6 Herpes simplex virus type 2 (HSV-2) is the cause of the most common sexually transmitted disease, and sporadic recurrent genital lesions occur periodically. Two genital herpes vaccine trials failed7,8 indicating there is a need for new and effective therapies that will reduce the incidence of recurrent HSV-1 and HSV-2 disease.
Bovine herpes virus 1 (BHV-1) is an important pathogen of cattle as it induces clinical signs in the upper respiratory tract of cattle and is immune-suppressive. BHV-1 establishes latency in sensory neurons, but periodically reactivates from latency, and thus is widespread in cattle.2,9–11 BHV-1 infection inhibits cell-mediated immunity, 12 15 CD8+ T cell recognition of infected cells, 16 19 and induces apoptosis in CD4+ T cells.20,21 Two viral regulatory proteins, bICP0 and bICP27, inhibit interferon dependent transcription.10,22–25 Infection also erodes mucosal surfaces of the upper respiratory tract, which can allow bacterial pathogens to colonize the lower respiratory tract. 26 28
Acute Infection Results in High Levels of Infectious Virus and Apoptosis
Binding and entry of HSV-1 and BHV-1 to mammalian cells are mediated by viral glycoproteins and cellular factors. 29 31 A cellular receptor (HveA or HVEM) is primarily expressed in activated T cells and belongs to the tumor necrosis factor receptor family. 32 Entry of HSV-1 into epithelial and fibroblasts is mediated by another membrane glycoprotein, HveB or HveC. 33 HveC is an entry mediator for HSV-1 and BHV-1 and is abundantly expressed in neurons. Additionally, soluble HveC blocks viral entry in neuronal-like cell lines. 33 After uncoating, the viral genome enters the nucleus and productive infection is initiated.
HSV-1 and BHV-1 gene expression is tightly regulated in three distinct phases during productive infection of cultured cells: immediate early (IE), early (E), or late (L). 34 IE RNA expression does not require protein synthesis and is stimulated by VP16, a viral structural protein. 35 E gene expression requires at least one IE protein, and E genes encode nonstructural proteins that stimulate viral DNA replication. L gene expression is maximal after viral DNA replication, requires IE protein production, and L proteins comprise the virion particle. Although a vigorous immune response leads to viral clearance following primary infection, BHV-1 and HSV-1 establish a life-long latent infection in ganglionic sensory neurons, primarily TG, or sacral dorsal root ganglia.2,3,9,36 Approximately 40% of sensory neurons appear to harbor viral genomes during latency. 37 41
Five HSV-1 IE genes encode ICP0, ICP4, ICP22, ICP27, or ICP47. ICP4 42 45 and ICP27 46 48 are required for virus growth in tissue culture. ICP4 represses IE gene expression44,49–53 but activates E or L gene expression by interacting with RNA polymerase II transcription factors and specifically binding viral DNA.49,54 ICP27 redistributes small nuclear ribonucleoprotein complexes, interferes with splicing of IE transcripts, and promotes E and L poly A site selection. 55 58 ICP47 prevents transport of antigenic peptides into the endoplasmic reticulum 59 and inhibits CD8+ T cell responses. 60 ICP22 enhances viral gene expression, in part by modifying RNA polymerase II. 61 ICP0 increases steady-state levels of viral mRNA and stimulates all viral promoters. 62 ICP0 also binds several cellular proteins: (1) elongation factor 1-α 63 ; (2) cyclin D3 64 ; (3) an ubiquitin-specific protease65,66; and (4) promyelocytic leukemia (PML) protein. 67 69 Interactions between ICP0 and chromatin-remodeling enzymes activate viral transcription by multiple mechanisms, including sequestering histone deacetylase (HDAC) inhibitors.70,71 Secondly, HSV-1 ICP0 interacts with HDAC2 72 and blocks histone deacetylation to stimulate viral gene expression.73,74 ICP0 also alters a complex that inhibits gene expression (REST/CoREST/HDAC repressor complex). 73 Since ICP0 can remove histones from viral chromatin during productive infection, 75 ICP0 may have similar functions during reactivation from latency. These activities of ICP0 promote virus replication in differentiated cells. 76 BHV-1 encoded ICP0 (bICP0) has similar functions as ICP0. 10
Viral infection routinely leads to apoptosis in cultured cells. 77 80 Killing of infected cells by apoptosis in vivo can reduce inflammation, alter immune recognition, reduce burst size, and thus prevent virus spread. Premature apoptosis of infected cells limits production of infectious virus and limits viral spread. Members of the α-herpesvirinae subfamily induce apoptosis after infection of cultured cells. 81 86 HSV-183,84,87–89 and BHV-1 90 can also inhibit apoptosis in a cell type dependent manner after infection of cultured cells. HSV can induce DNA damage, and consequently apoptosis, even in the absence of productive infection. 91 95 Two viral proteins, US1.5 and UL13, activate caspase 3 in the absence of other viral proteins, indicating these viral proteins play an important role during virus mediated apoptosis. 96 Finally, ICP0 is also a trigger for apoptosis in the context of productive infection, in part because it activates viral gene expression.
HSV-1 encodes several proteins (ICP27, Us3, Us5, gJ, gD, and LAT) that have anti-apoptosis activity.83–85,87,88,97–105 Us3 is a serine/threonine protein kinase that inhibits cleavage and activation of the pro-apoptotic Bcl-2 family member, Bad. Us3 protein expression in cultured cells, in the absence of other viral proteins, inhibits caspase 3 activation, a crucial executioner caspase that commits cells to apoptosis. As expected, US3 inhibits the pro-apoptotic activity of US1.5 and UL13 by blocking caspase 3 activation. 96 These anti-apoptotic genes play an important role in the pathogenic properties of HSV-1.
Viral Genes Expressed during Latency Regulate the Latency-Reactivation cycle
The HSV-1 Latency Associated Transcript is Abundantly Expressed during Latency and Regulates the Latency-Reactivation Cycle
The HSV-1 latency associated transcript (LAT) is abundantly expressed in sensory gangionic neurons of mice, rabbits, or humans that are latently infected. 106 114 LAT is predominantly expressed in the nucleus of latently infected neurons suggesting it is a non-protein coding regulatory RNA. LAT is antisense to ICP0 and overlaps ICP0 mRNA sequences (Fig. 1B), suggesting LAT inhibits ICP0 expression by an anti-sense mechanism. Although the ability of LAT to repress ICP0 expression may be important, LAT sequences that promote spontaneous reactivation in a rabbit ocular model of infection do not overlap ICP0 mRNA sequences. 115
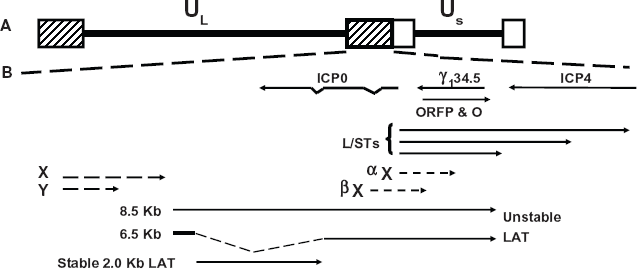
Location of genes within the HSV-1 repeats. (Panel
Splicing of the 8.5 kb LAT transcript yields a sTable 2 kb LAT and an unsTable 6.5 kb LAT (Fig. 1B).107,111,116 Correct splicing of the 2 kb LAT is necessary for establishment and maintenance of latency.117,118 In general, the 2 kb LAT is not capped, is poly A-, appears to be circular, and is a stable intron.119,120 A subset of LAT is detected in the cyto-plasm97,121,122 and is associated with polyribosomes or splicing factors.97,123 Small non-coding RNAs can regulate gene expression,124,125 promote neuronal differentiation, 126 or inhibit apoptosis 127 suggesting LAT is a non-coding regulatory RNA.
A study by Umbach et al 128 concluded that LAT is a microRNA (miRNA) precursor which encodes four miRNAs, two within LAT promoter sequences (Fig. 2A and B). LAT miR-H6, reduces ICP4 protein steady state levels but not ICP4 RNA levels. Protein levels, not RNA levels, of ICP0 are inhibited by the LAT miRNA, miR-H2-3p. Within the first 1.5 kb of LAT coding sequences, two small RNAs (sRNAs), LAT sRNA1 and sRNA2, were identified (Fig. 2B). The sRNAs are larger than mature miRNAs (typically 23 nucleotides long) and the sequence of both possess extensive secondary structure. The sRNAs can be detected in TG of mice latently infected with wild-type HSV-1, but not in TG of mice latently infected with a LAT null mutant.79,129 LAT sRNA2, but not LAT sRNA1, reduced ICP4 protein levels in transient transfection assays. Both LAT sRNAs inhibit productive infection in mouse neuroblastoma cells, however LAT sRNA1 inhibited productive infection more efficiently than LAT sRNA2. 130 Collectively, these studies provide evidence that the LAT encoded miR-NAs and sRNAs promote latency by interfering with expression of important viral transcriptional regulatory proteins.
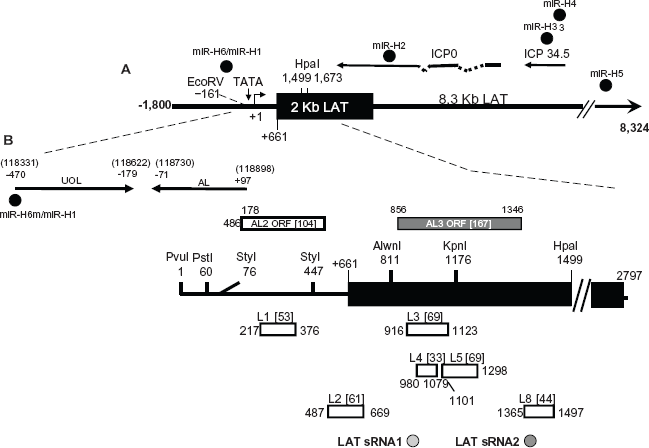
Schematic of putative factors encoded within the LAT locus. (Panel
The LAT locus encodes additional transcripts (Fig. 2B). More than one transcript, including UOL (Upstream of LAT) 131 are located within LAT promoter sequences. Expression of the UOL transcript or protein does not reduce reactivation from latency in rabbits. 132 An antisense to LAT (AL) transcript is expressed from the first 1.5 kb of LAT coding sequences and appears to encode a protein. 133 Two additional small open reading frames (ORFs) that are antisense to LAT (AL2 and AL3) are present in LAT coding sequences. An AL3 transcript is expressed during productive infection and in TG of mice latently infected with wild-type HSV-1, but not a LAT null mutant virus. 134 An AL3-specific polyclonal antibody detected a protein in a subset of TG neurons in latently infected mice. A transcript encompassing AL2 has not been detected during productive infection or latency (unpublished data).
LAT null HSV-1 mutants have been examined in various small animal models.2,3 Although two studies concluded that LAT does not play a role in latency,135,136 most have provided evidence that LAT is important. This discrepancy may be due to the strain of virus or mouse that was used for specific studies. LAT enhances the establishment of latency in mice137,138 and in rabbit ocular infection models, 139 in part by reducing lytic cycle viral gene expression in TG of mice.140,141 By enhancing the establishment of latency, LAT would increase the pool of latently infected neurons; thus indirectly increasing the incidence of reactivation from latency.
As a result of stress or other external stimuli, reactivation from latency can occur, resulting in virus shedding (Fig. 4). The McKrae strain of wild-type HSV-1, however not a LAT null mutant, is consistently detected in tears of infected rabbits, due to spontaneous reactivation.139,142–145 These same LAT null mutants grow with wild-type efficiency in cultured cells and in acutely infected rabbits. When just the first 1.5 kb of LAT coding sequences (Fig. 2B) is expressed from the HSV-1 genome, wild-type levels of spontaneous reactivation from latency occur in rabbits. 139 Similar results were observed using another virulent strain of HSV-1 (17 syn+) in a rabbit eye model.146,147 The first 1.5 kb of LAT coding sequences does not overlap ICP0, suggesting that antisense repression of ICP0 expression by LAT is not important for spontaneous reactivation in the rabbit ocular model of infection. The factors encoded by the first 1.5 kb of LAT coding sequences that promote spontaneous reactivation have not been fully characterized.
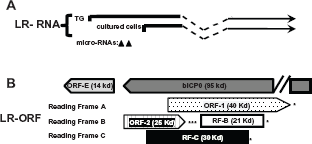
Schematic of the BHv-1 LR gene and surrounding genes. (Panel
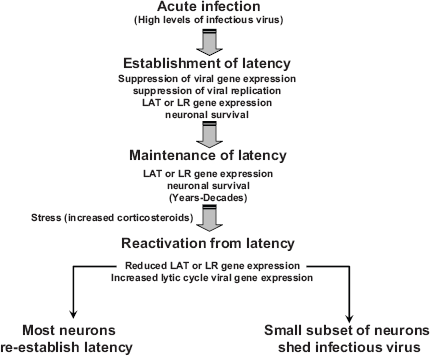
Putative steps that occur during the latency-reactivation cycle.
Although certain studies suggested LAT does not encode a protein, 148 other studies have concluded that a protein encoded within LAT sequences is expressed.131,149–154 These proteins were suggested to either substitute for ICP0 functions,153,154 interfere with binding of ICP4 to DNA, 152 or their functions were not described. The proposed LAT proteins are mapped downstream of the critical first 1.5 kb of the primary LAT transcript, a region that appears both sufficient and necessary for the wild type spontaneous reactivation phenotype in rabbit models.139,155 Within the first 1.5 kb of LAT coding sequences, 8 potential ORFs have been identified in the McKrae strain (Fig. 2B). 148 The L2 ORF (Fig. 2B) appears to be expressed in TG of latently infected mice. 156 Although LAT is not absolutely required for the latency-reactivation cycle in small animal models, its importance may be underestimated using small animal models and measuring latency in terms of weeks or months, not decades.
The BHV-1 Latency Related RNA is Abundantly Expressed in Sensory Neurons and is Necessary for Reactivation from Latency
Latency related (LR) RNA is abundantly expressed in TG neurons of calves that are latently infected.111,157 Two different start sites of LR-RNA transcription (Fig. 3A) have been identifed suggesting this has functional significane. It is clear that the LR gene encodes more than one product.11,36 For example, the LR gene contains two-well defined ORFs (ORF2 and ORF1; Fig. 3B) and two reading frames that lack an initiating methionine (RF-B and RF-C). As a result of alternative splicing of polyA+ LR-RNA in TG of infected calves (Fig. 3A),158,159 ORF2 can be fused with ORF1 protein coding sequences or RF-B. The ORF2/ORF1 fusion protein stably interacts with the cellular transcription factor C/EBP-alpha. 160 C/EBP-alpha RNA and protein levels increase in TG neurons during dexamethasone induced reactivation from latency. Over-expression of C/EBP-alpha enhanced productive infection, 161 suggesting that ORF2 sequesters C/EBP-alpha and reduces the efficieny of productive infection during the latency-reactivation cycle.
One day after calves are infected and during latency, splicing of LR-RNA in TG is such that ORF2 is intact, 159 suggesting ORF2 expression is important for the latency-reactivation cycle. ORF2 interacts with Notch1 and Notch3, components of the Notch signaling pathway. 162 Mammalian Notch receptor family members (Notch1-4) are membrane tethered transcription factors that regulate many developmental and physiological processes.163,164 For example, Notch promotes neuronal maintenance, development, and differentiation. 165 167 Notch3 168 and Notch1169,170 promote cell survival by activating a protein kinase, (AKT) which inhibits apoptosis. Notch family members can also induce apoptosis,163,164 suggesting Notch influences cell survival by cell-type dependent mechanisms. When the Notch receptor is engaged by one of its five transmembrane ligands (Jagged1, Jagged2, Delta-like1, Delta-like3, or Delta-like4), the Notch intracellular domain (ICD) is cleaved by specific proteases, and subsequently translocates to the nucleus. In the nucelus, Notch ICD interacts with members of the CSL family of transcriptional factors, CBF1, Su(H), or Lag1 (also referred to as RBP-J binding proteins) subsequently activating downstream genes. Notch1, but not Notch3, enhances BHV-1 productive infection 163 and Notch1 activates the BHV-1 immediate-early transcription unit 1 (IEtu1) and bICP0 early promoters. Notch1 and Notch3 trans-activated the late glycoprotein C (gC) promoter. ORF2 interferes with the ability of Notch1 to trans-activate the bICPO early promoter and Notch1 or Notch3 mediated activation of the gC promoter 162 suggesting this function is important for establishing and/or maintaining latency. Notch3 RNA levels are higher during dexamethasone (DEX) induced reactivation from latency, suggesting Notch family members stimulate productive infection during reactivation from latency. Activation of Notch signaling in post-mitotic-neurons or neuroblastoma cells inhibits neurite sprouting165,171–174 and axon repair, 175 which can lead to neuronal degeneration and apoptosis. 176 178 Conversely, neurite sprouting correlates with regeneration of damaged axons and dendrites. 175 ORF2 promotes neuruite sprouting and neuronal differentiation of mouse neuroblastoma cells when Notch1 or Notch3 is over-expressed. 179 Collectively, these studies suggest that ORF2 interactions with Notch family members promote the establishment and maintenace of latency by (1) interfering with viral gene expression necessary for productive infection, (2) supporting a mature neuronal phenotype, and (3) overcoming the deleterious effects of Notch expression during stress-induced reactivation from latency.
Although the results from the LR mutant virus suggested that proteins encoded by the LR gene are necessary for the latency-reactivation cycle, non-protein coding functions within LR-RNA have also been identified. For example, the intact LR gene inhibits the ability of bICP0 to stimulate productive infection in a dose-dependent manner.180,181 Insertion of three in-frame stop codons at the amino-terminus of the first ORF within the LR gene (ORF2) inhibited bICP0 repression with similar efficiency as the wild-type LR gene, suggesting expression of a LR protein is not required. 181 LR gene products also inhibit mammalian cell growth,182,183 and the cell growth inhibitory function of the LR gene maps to a 463-bp fragment that lacks a significant open reading frame. 182 Two miR-NAs located upstream of ORF2 are expressed during latency. 184 These miRNAs, or larger sRNAs containing these miRNAs, reduced bICP0 protein levels in transient transfection assays.
A small ORF located within the LR promoter is designated ORF-E (Fig. 3B). ORF-E is antisense to the LR transcript and is downstream of bICPO coding sequences, but does not overlap bICP0. A transcript that encompasses ORF-E is expressed during productive infection and in TG of latently infected calves. 185 The LR promoter contains multiple cis-acting motifs, has a neuronal specific binding domain, 186 188 and contains a long AT-rich motif (40/53 nucleotides are A or T) that may promote ORF-E transcription. When ORF-E protein coding sequences are fused in frame with green fluorescent protein (GFP) sequences, GFP protein expression is detected in the nucleus of mouse or human neuroblastoma cells. In contrast, the ORF-E-GFP fusion protein is detected throughout rabbit skin cells. In transient transfection assays, ORF-E promotes neurite formation in mouse neuroblastoma cells, 189 which may support a mature neuronal pheno-type following infection.
LAT and LR Gene Products Inhibit Apoptosis
LAT Inhibits Apoptosis
LAT expressing plasmids interfere with apoptosis in transiently transfected cells, and LAT expressing viruses inhibit apoptosis in TG of infected mice or rabbits.117,190–192 The anti-apoptotic functions of LAT correlate with promoting spontaneous reactivation from latency.191,193 In the context of promoting spontaneous reactivation from latency in the rabbit model (model), inhibiting apoptosis is the most important function of LAT as three different anti-apoptosis genes129,194–196 restore wild-type levels of spontaneous reactivation from latency to a LAT null mutant. LAT may encode other functions because the LAT null mutants that express cellular anti-apoptosis genes have reduced virulence, in spite of reactivating from latency with wild-type frequency. LAT expressing plasmids, in the absence of other viral genes, inhibit caspase 8- and caspase 9-induced apoptosis,193,197 the two major apoptotic pathways in mammals. 198 200 LAT also inhibits caspase 3 activation. 201
LAT sRNA1 and sRNA2 cooperate to inhibit cold-shock induced apoptosis in mouse neuroblastoma cells. 130 Introduction of ATG→TTG mutations in ORFs within the first 1.5 kb of LAT coding sequences impairs the anti-apoptotic functions of LAT, 202 suggesting that LAT either encodes a functional protein or alters RNA structure. Two of these ATG→TTG mutations are within LAT sRNA1 and sRNA2, and introducing these mutations into the small RNAs inhibits their ability to inhibit apoptosis. 130 At this time, it is not clear how these sRNAs interfere with apoptosis. It will also be important to construct a recombinant virus with these same mutations and test whether the spontaneous reactivation incidence is affected.
LAT also inhibits GrzB induced apoptosis in transient transfection studies. 203 GrzB is released from CD8+ T cells as well as other specific lymphocytes; GrzB has features similar to apical caspases, and can induce apoptosis in most cell types. 204 207 Inhibiting GrzB induced apoptosis may be important for the latency-reactivation cycle because CD8+ T lymphocytes control HSV infection in sensory ganglia.208,209
The LR Gene Encodes More than One Product that Inhibits Apoptosis
A mutant BHV-1 strain with 3 stop codons after the initiating methionine codon of ORF-2 (LR mutant virus) does not express detectable levels of ORF-2 101 but expresses reduced levels of ORF1 in cultured cells during productive infection. 210 The LR mutant virus grows less efficiently in the ocular cavity and TG, but grows almost as efficiently as wild-type BHV-1 in the nasal cavity, and does not reactivate from latency following DEX treatment.211,212 The LR mutant virus induces higher levels of apoptosis in TG neurons of infected calves, 213 and a LR gene expressing plasmid with the same stop codon mutations does not effectively inhibit apoptosis.214,215 ORF2 expression in the absence of other viral genes inhibits apoptosis in transiently transfected cells,216,217 suggesting that ORF2 is a dominant function encoded by the LR gene. ORF2, like LAT, can inhibit caspase 8 and caspase 9 mediated apoptosis; however the mechanism by which it inhibits apoptosis is not known.
Two microRNAs encoded within the LR gene (Fig. 3A) interfere with bICP0 protein expression 184 and cold shock induced apoptosis in transfected mouse neuroblastoma cells (Neuro-2A cells). 218 Since cold shock induced apoptosis in Neuro-2A cells is inhibited by casapse 3 and caspase 9 inhibitors, 219 the microRNAs must influence these apoptotic signaling pathways. The ability of the microRNAs to stimulate the anti-apoptotic transcription factor NF-αB 220 223 seems to be important for inhibiting cold-shock induced apoptosis. In summary, these results provide additional evidence that interfering with apoptosis is crucial for a successful life-long latent infection.
Why is Inhibiting Neuronal Apoptosis Important during the Latency-Reactivation Cycle?
The latency-reactivation cycle has been operationally divided into three distinct steps: establishment, maintenance, and reactivation (Fig. 4). Following acute infection where high levels of infectious virus are produced, virus particles enter sensory neurons. Initial entry of the viral genome into a sensory neuron results in a burst of lytic cycle viral gene expression and infectious viruses are produced. Viral gene expression is then extinguished, with the exception of HSV-1 LAT and BHV-1 LR gene products. Neuronal cell factors,2,9 LAT encoded microRNAs plus sRNAs,128,130 and LR encoded functions162,184 interfere with various aspects of productive infection. During acute infection and establishment of latency, neuronal and satellite cells undergo apoptosis when small animal models are infected with HSV-2 224 or HSV-1. 225 227 BHV-1 replication and gene expression also occur in TG of acutely infected calves, resulting in apoptosis of neurons and non-neuronal cells.192,213,228
HSV-1 LAT 192 and the LR gene 213 enhance neuronal survival during the establishment of latency. The ability of LAT 229 and the LR gene 179 to promote a mature neuronal phenotype and sprout neurites may also promote establishment of latency by stimulating repair of damaged neurons following infection. Successful establishment correlates with an increase in the number of infected neurons that survive and enhances the probability that reactivation from latency occurs.
Maintenance of latency is a phase that lasts for the duration of the host's life and is operationally defined as a period when infectious virus is not readily detected. In general, abundant expression of viral genes required for productive infection does not occur. LAT or LR gene products are abundantly expressed during the maintenance of latency. Expression of LAT correlates with an increase of latently infected neurons during the maintenance of latency, 230 suggesting latently infected sensory neurons are exposed to apoptotic stimuli during the maintenance of latency. It is reasonable to predict that LAT and LR gene products actively participate in maintaining a latent infection in sensory neurons.
Reactivation from latency is initiated by external stimuli (stress, immunosuppression, or UV light for example), which ultimately must stimulate viral gene expression.36,231,232 Abundant viral gene expression can be detected in sensory neurons and infectious virus can be isolated from TG, ocular swabs, and/or nasal swabs. Stress leads to elevated corticosteriod levels, which has rapid effects on neural activity.233,234 DEX, a synthetic corticosteriod, induces viral gene expression, 235 stimulates an HSV-1 origin of replication (Ori-L) in neuronal cells, 55 and alters splicing patterns in the absence of protein synthesis. 236 DEX and other apoptosis stimulators can also stimulate HSV-1 reactivation from latency.237,238 BHV-1 reactivation from latency is induced by DEX, in part because it stimulates expression of cellular transcription factors and viral gene expression while repressing expression of LR gene products.9,11,36 Prolonged exposure to corticosteroids can also induce immunosuppression, in part by inducing apoptosis in lymphocytes. 239 A subset of neurons that successfully reactivate from latency to produce infectious virus may not survive; 240 242 however it is not clear if this is the fate for all neurons that produce infectious virus during reactivation from latency. Most latently infected neurons that are exposed to reactivation stimuli re-establish latency and do not produce infectious virus.243,244 Given that sensory neurons are terminally differentiated cells, inhibiting apoptosis during the latency-reactivation cycle is crucial for life-long latent infections of α-herpesvirinae subfamily members.
Numerous studies have demonstrated that infiltrating lymphocytes in TG regulate the latency-reactivation cycle. For example, a persistent cell-mediated immune response occurs in TG during latency and CD8+ T lymphocytes inhibit reactivation from latency.208,209,245–250 Release of granzyme B from CD8+ T cells into latently infected neurons helps to inhibit reactivation from latency by cleaving the viral transcriptional trans-activator, ICP4. 251 Since it is well established that granzyme B activates caspase 3 and the intrinsic pathway of apoptosis, 207 the ability of LAT and perhaps LR gene products to inhibit apoptosis is important to overcome the effects of granzyme B. The ability of HSV-1 to inhibit major histocompatibility complex (MHC) class I presentation in sensory neurons correlates with successful reactivation 252 providing further evidence that CD8+ T cells monitor latently infected neurons. In conclusion, the ability of HSV-1, HSV-2, and BHV-1 to reactivate from latency is regulated by complex virus-host interactions.
Perspectives
Although genetic and functional studies have demonstrated that LAT and the LR gene regulate the latency-reactivation cycle, there are many unanswered questions. For example, identifying the functions of various transcripts, the small non-coding RNAs, and the ORF s within LAT are crucial to understand the role these various factors play in the latency-reactivation cycle. It would not be surprising to find that one or more of these factors encoded within the LAT locus regulate certain neuronal specific functions that maintain normal functions. It will be difficult to make additional LAT mutant viruses as many of these factors overlap and deletion of these sequences would likely interfere with expression of more than one LAT encoded factor. Consequently, many of these studies will have to be performed in transient transfection assays in primary neurons or neuroblastoma cells. Finally, examining LAT in small animal models in terms of weeks after acute infection may not accurately reflect the latency-reactivation cycle in the context of life-long latency in humans.
With respect to the LR gene, there are no studies that have determined whether ORF1, ORF-E, or the microR-NAs play a role in the latency-reactivation cycle. Furthermore, functional analysis of ORF-1 and ORF-E has not been performed. Identifying the cellular proteins that interact with ORF-1 and ORF-E may provide insight into their functions. In summary, the finding that sequences encompassing LAT and the LR gene encode for more than one transcript and/or small non-coding RNAs implies many functions are necessary to successfully regulate the lifelong latency-reactivation in the natural host.
Author Contributions
Conceived and designed the experiments: CJ. Analyzed the data: CJ. Wrote the first draft of the manuscript: CJ. Contributed to the writing of the manuscript: CJ. Agree with manuscript results and conclusions: CJ. Jointly developed the structure and arguments for the paper: CJ. Made critical revisions and approved final version: CJ. All authors reviewed and approved of the final manuscript.
Funding
Research in the author's laboratory is supported by a grant from the USDA, NIFA Competitive Grants Program (09-01653), a grant to the Nebraska Center for Virology (1P20RR15635), and the NE Research Initiative.
Competing Interests
Author disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the author has provided signed confirmation of compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests. Provenance: the authors were invited to submit this paper.