
Abstract
Efficient synthesis of NAD+ is critical to maintaining cell viability in all organs of the body. However, little is known of the pathway(s) by which cells of the central nervous system produce NAD+. The aim of this study was to investigate the relationship, between tryptophan degradation via the kynurenine pathway (KP) and
Introduction
Nicotinamide adenine dinucleotide (NAD+), as the parent molecule for the pyridine family of nucleotides, (NADH, NADP and NADPH) is an important coenzyme enabling electron transfer in the oxidative production of ATP and for hydride ion transfer in many enzyme reactions. Depleted mitochondrial NAD+ impairs respiration and ATP synthesis resulting in energy crisis and cell death.
1
More recently NAD+ has also been identified as a primary substrate for other important enzymes including poly (ADP-ribose) polymerase (PARP) and the sirtuin family of

Schematic of NAD+ Biosynthesis;
The aim of this study was therefore to investigate the relationship, between kynurenine pathway (KP) metabolism and
Materials and Methods
Reagents and chemicals
All cell culture media and supplements were purchased from Invitrogen (Australia) unless otherwise stated. All reagents and chemicals used in experiments were purchased from Sigma Aldrich Chemical Co. (Australia) unless otherwise stated.
Cell cultures
Human primary astrocytes were grown in uncoated flasks (Falcon) and maintained in complete media (cRPMI), which contains RPMI 1640 media supplemented with 10% foetal bovine serum (FBS), 1% 2 mM glutamine (Sigma Aldrich, Australia) and 1% penicillin/streptomycin (Sigma Aldrich, Australia). As part of its normal commercial formulation cRPMI contains 24 μM tryptophan and 8 μM nicotinamide. The cell medium was changed twice a week, and all cell cultures were kept incubated at 37 °C in 5% CO2. 9
A day prior to experimental treatments, cultures were trypsinised (Trypsin 0.25%) and seeded at desired cell density, into 24 well plates (Falcon).
NAD+ precursor depleted RPMI
Depleted RPMI (dRPMI) was prepared using a standard mix formula for RPMI that included all nutrients except tryptophan (TRYP) and nicotinamide (NAM). Note that standard RPMI does not contain nicotinic acid (NIC) or kynurenine (KYN).
The dRPMI medium containing 10% FBS, 1% 2 mM glutamine (Sigma Aldrich, Australia) and 1% penicillin/streptomycin (Sigma Aldrich, Australia) was supplemented as required with the addition of substrates TRYP (25 μM), KYN (25 μM) NAM (10 μM) or NIC (10 μM). The concentrations chosen for TRYP and NAM supplementation correspond to that found in the commercially prepared RPMI (cRPMI). As KYN is not present in RPMI media this upstream metabolite was added at a concentration comparable to TRYP, which has been shown to increase in the culture medium of stimulated astroglial cells. 13 NIC (the acid form of vitamin B3) is also not normally present in RPMI media and was added at the same concentration as its amide form, NAM to allow direct comparison. All supplements were prepared as sterile solutions in purified (>10 mω) water).
NAD+ precursor supplementation
Astrocytes were seeded into a 24-well culture plate at a density of 1 × 105 cells/ml in and left to equilibrate in 5% CO2 at 37 °C in complete RPMI (cRPMI) for 24 hr. The culture medium was then aspirated and each well was washed twice with warm phosphate buffered saline (PBS) before addition of 1 ml of fresh dRPMI (containing no TRYP, NAM or NIC). Selected NAD+ substrates TRYP (25 μM), KYN (25 μM) NAM (10 μM) or NIC (10 μM) were then added to appropriate cultures for 24 hrs before analysis.
Kp enzyme inhibition
Human primary astrocytes were plated into 24-well plates at a density of 1 × 105 cells/ml in cRPMI and left to equilibrate for 24 hours before treatment. Cultured medium was aspirated from each well, washed twice with warm phosphate buffered saline (PBS), before addition of 1 ml of fresh cRPMI followed by addition of specific inhibitors (Table 1) for 24 hrs.
Inhibitors used in the experiments.
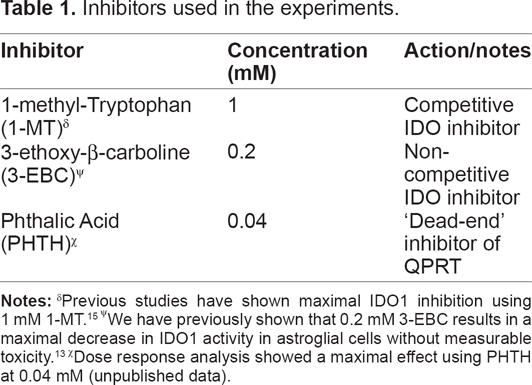
We have previously shown that 0.2 mM 3-EBC results in a maximal decrease in IDO1 activity in astroglial cells without measurable toxicity. 13
Dose response analysis showed a maximal effect using PHTH at 0.04 mM (unpublished data).
NAD+ analysis
Cultured medium was aspirated from each well. The cells were washed twice with PBS, before being homogenised by sonication in 500 μl of PBS (containing 12 mM nicotinamide as a PARP inhibitor). Quantification of NAD+ (H) in the cell was measured using the Thiazolyl blue microcycling assay 11 adapted to a 96-well format. 7
Total protein assay
Cultured medium was removed from each well and the cells were washed twice with PBS, before being homogenised by sonication in 500 μl of PBS (containing 12 mM nicotinamide as a PARP inhibitor). The total protein per sample was measured using the commercially available Bradford assay method and reagents (BIORAD, Sydney, Australia), adapted to a 96-well microtitre plate format, using a 595 nm filter in a Multiskan MS microplate reader.
Statistical analysis
For all culture samples, significant differences between treatment groups, at
Results
Involvement of KP enzymes in de novo NAD+ synthesis
To determine whether tryptophan catabolism via the KP played a role in NAD+ synthesis, we used small molecule inhibitors that reduced the activity of critical enzymes at the beginning and end of the KP.
Inhibition of the initial rate limiting enzyme indoleamine 2,3 dioxygenase (IDO1) with either the competitive inhibitor, 1-methyl tryptophan (1-MT) or the non-competitive inhibitor, 3-ethoxy-β-carboline (3EBC) significantly decreased intracellular NAD+ by 14% and 26% respectively after 24 hrs. In a similar fashion, inhibition of the downstream KP enzyme, quinolinic acid phosphoribosyl transferase (QPRT) with the dead-end inhibitor phthalic acid (PHTH) resulted in a significant 29% decrease in intracellular NAD+ levels after 24 hrs (Fig. 2).

Effect of inhibition of selected KP enzymes on NAD+ levels in primary human astrocytes. Cellular NAD+ levels were significantly reduced in primary human astrocyte cultures (1 × 105 cells/ml) following treatment with: The competitive IDO inhibitor 1-methyl tryptophan (1-MT 1 mM) or the non-competitive IDO inhibitor, 3-ethoxy, β carboline (3-EBC, 0.2 mM) or the QPRT inhibitor phthalic acid (PHTH, 0.04 mM) for 24 hrs.
Importance of de novo and salvage pathway precursors to NAD+ synthesis
To help verify that the results obtained for enzyme inhibition were specific to KP metabolism, we tested whether the removal of KP or salvage pathway precursors (i.e. tryptophan, nicotinic acid and nicotinamide) from the incubation medium affected cellular NAD+ levels.
We found that in the absence of all of these precursors a significant decrease of 50% in intracellular NAD+ levels after 24 hrs was observed (Fig. 3).
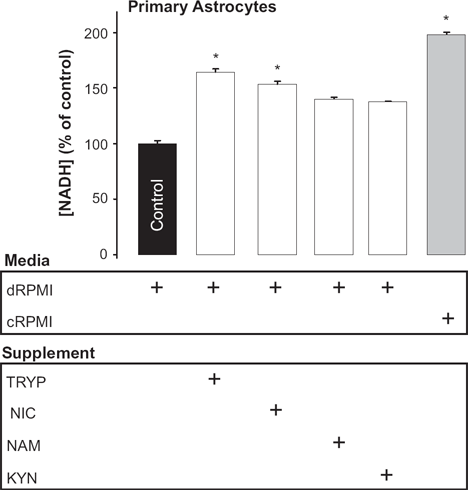
Effect of substrate availability on cellular NAD+ levels in primary human astrocytes. Primary human astrocytes grown in either dRPMI (RPMI with 10% FBS without the NAD+ precursors tryptophan, nicotinic acid or nicotinamide) or cRPMI (complete RPMI with 10% FBS containing tryptophan and nicotinamide) ± addition of the specific NAD+ precursors tryptophan (TRYP, 25 μM), nicotinic acid, (NIC, 10 μM), nicotinamide (NAM, 10 μM) or kynurenine (KYN, 25 μM) for 24 hrs resulted in a significant increase in NAD+ levels.
Subsequent supplementation of the incubation medium with the upstream KP precursor tryptophan alone, increased NAD+ levels by 75% within 24 hrs compared to control. Addition of nicotinic acid to the culture medium also increased NAD+ levels significantly by 54% after 24 hrs (Fig. 3). Though an apparent increase in NAD+ was observed with kynurenine and nicotinamide supplementation, these did not reach statistical significance (Fig. 3).
Discussion
We have previously shown that KP metabolism is linked to the maintenance of NAD+ levels in murine primary astrocytes and human and murine cell lines.7,12,13 However, following the observation that primary human astrocytes do not express the KP enzyme, kynurenine 3-hydroxylase, 9 we were unsure whether these cells could efficiently use tryptophan for NAD+ synthesis. Using the dual approach of enzyme inhibition and NAD+ substrate depletion, we investigated the role of the KP and salvage pathway metabolism in the maintenance of NAD+ concentrations in primary human astrocytes.
In the present study, we observed that intracellular NAD+ levels decreased significantly following inhibition of either the initial or downstream KP enzymes IDO1 or QPRT, respectively (Fig. 2). These results are consistent with our previous findings showing a clear relationship between IDO activity and NAD+ levels in a human astroglioma cell line. 13 Supporting these results, we also observed that intracellular NAD+ levels decreased significantly after 24 hrs in the absence of either KP metabolites or salvage pathway substrates in the incubation medium (Fig. 3).
The subsequent regeneration of NAD+ from TRYP, KYN, NIC or NAM (Fig. 3) strongly suggests the metabolic use of these precursors in the production of NAD+.
This result is similar to our previous reports showing that quinolinic acid or nicotinic acid or nicotinamide were able to regenerate NAD+ after oxidative stress in murine primary astrocytes 7 . In the present study the observation that primary human astrocytes regenerated NAD+ more efficiently from NIC than NAM when supplied in equimolar concentrations (Fig. 3) is indicative of preferred metabolism from this source. This is again consistent with our previous report in murine primary astrocytes showing NIC was also more efficient at promoting NAD+ regeneration than the amide. 7
Taken together these results support the hypothesis that the KP is an important route for NAD+ synthesis in primary human astroglial cells. As this occurs in spite of the previously reported absence of kynurenine 3-hydroxylase, 9 the progress around this blockade to NAD+ most likely occurs via efficient non-enzymatic hydroxylation of anthranilic acid to 3-hydroxyanthranilic acid (Fig. 1). This is consistent with studies done some time ago by Henderson et al who reported that kynurenine could be converted to niacin (NAD+) in kidney and liver tissue through non-enzymatic hydroxylation. 16
Importantly, though tryptophan catabolism via the KP clearly results in NAD+ production, significant synthetic capacity is also possible via nicotinic acid or nicotinamide and the salvage pathway (Fig. 3).
With growing interest in IDO1 inhibition as a therapeutic target 14 coupled with the fact that maintaining NAD+ levels is an essential part of organ health 10 characterisation of NAD+ metabolism in other cells of the CNS such as neurons and microglia are needed. Further research is also required to confirm whether NAD+ levels can be effectively maintained when IDO1 or other KP enzymes are inhibited by concurrent addition of NIC or salvage pathway precursors such NAM.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Acknowledgements
This study was supported by a research grant from the Australasian Research Institute.