
Abstract
Autism spectrum disorder (ASD) is a pervasive neuro-developmental disorder characterized by impaired social interaction, reduced/absent verbal and non-verbal communication, and repetitive behavior during early childhood. The etiology of this developmental disorder is poorly understood, and no biomarkers have been identified. Identification of novel biochemical markers related to autism would be advantageous for earlier clinical diagnosis and intervention. Studies suggest that oxidative stress-induced mechanisms and reduced antioxidant defense, mitochondrial dysfunction, and impaired energy metabolism (NAD+, NADH, ATP, pyruvate, and lactate), are major causes of ASD. This review provides renewed insight regarding current autism research related to oxidative stress, mitochondrial dysfunction, and altered tryptophan metabolism in ASD.
Introduction
Autism/autistic spectrum disorders (ASD) are lifelong neurodevelopmental disorders diagnosed within the first 3 years of life. Kanner 1 described autism in 11 children manifesting symptoms of withdrawal from human contact as early as age 1 year and postulated the origins of ASD in prenatal stages. ASD is a heterogeneous disorder, both etiologically and phenotypically. The Diagnostic and Statistical Manual of Mental Disorders IV-Text Revised (DSM-IV-TR) described autism as a complex neurobehavioral disorder characterized by varying degrees of social interaction deficits, flexibility of thinking, specific language abnormalities and/or delayed cognition, impaired verbal and non-verbal communication, failure to respond to certain stimuli with some restricted and extreme repetitive behavior or actions, stereotyped patterns of behavior or circumscribed interests, and association with several metabolic disorders. In recent years, autism has been shown to affect neural circuitry before 3 years of age. ASDs, encompassing full-syndrome autism (autistic disorder), Asperger syndrome, tuberous sclerosis, Angel man syndrome, congenital rubella, Down syndrome, Rett syndrome, asphyxia, childhood disintegrative disorder, and pervasive developmental disorder not otherwise specified (PDDNOS), are now grouped as ASDs, also known as pervasive developmental disorders (PDDs).2–6 Motor abnormalities in ASD can be observed in infancy, 7 and are apparent throughout childhood and into adulthood.8,9
The prevalence of autism has increased by more than tenfold in the last decade. Epidemiological studies conducted in several states of the USA, the United Kingdom, Europe, and Asia, estimate that 3.4 of every 10,000 children 3–10 years old will have autism. 10 The prevalence of ASDs in Saudi Arabia is 6:1000, 11 with a male to female ratio of 4.2:1. 12 Rates in the United States have increased from less than 3 per 10,000 children in the 1970s to between 34 and 93 children per 10,000 in the 2000s.13,14 The incidence of ASD was only 1.4 cases in 10,000 Omani children in the year 2011.16,17 A recent study in South Korea demonstrated a considerably higher figure of approximately 1 in 38 (ie, 260 out of 10,000) children diagnosed with ASD. 18 In Western Australia, the prevalence of ASD increased from 0.8/1000 in 1983 to 4.6/1000 in 1999, while this ratio increased from 6.6/1000 in 2000 to 9/1000 in 2006 in the United States. 19 This growing prevalence will have enormous future public health implications and has stimulated intense research into potential etiologic factors.
Despite expanding research regarding ASD, little is known regarding the pathology and etiology of these disorders; however, the relative contribution of genetic, epigenetic, immune response, nutrient, toxins, environmental, bowel dysfunctions, inflammatory, and environmental susceptibility factors, as well as increased vulnerability to oxidative stress has been examined.20–25 Environmental factors, such as mercury, lead, measles, rubella virus, zinc, retinoic acid, maternal thalidomide, valproic acid, and alcohol use during pregnancy have been suggested to be involved in the etiology of autism.26–29 Autistic persons have a high prevalence of gastrointestinal disease and dysbiosis, 30 epilepsy, 31 autoimmune disease, 32 and mental retardation 33 have also been indicated in the etiology of autism. Several studies have shown that an abnormal immune response may contribute to the etiology of certain forms of autism, including polymorphisms in immune genes controlling and regulating immune cell function, microglia and astroglia activation, production of proinflammatory cytokines, increased presence of central nervous system reactive antibodies, T cell activation, and innate immune activation. 34 Prenatal environmental factors, such as obstetrical suboptimality, alcohol exposure, and intrauterine infections, have also been reported to influence the occurrence of autism. 35 In addition, emerging evidence points to inflammatory and apoptotic mechanisms as responsible for certain neuropsychiatric disorders, including autism. Vargas et al 36 suggested that neuroinflammatory processes occur in autistic brains by showing that transforming growth factor-α1, macrophage chemo attractant protein 1, interleukin (IL)6, and IL10 are increased in the brain of autistic subjects. A number of studies have also shown that inflammatory cytokines, including tumor necrosis factor (TNF)-α, interferon-α, IL1α, IL6, IL8, and IL12, are elevated in blood mononuclear cells, serum, plasma, and cerebrospinal fluid of autistic subjects.20,36–40 Dimayuga et al 41 showed that overexpression of superoxide dismutase 1 (SOD1) in microglial cells leads to a significant reduction in superoxide concentrations, with corresponding increases in H2O2 concentrations. They further showed that the release of the proinflammatory cytokines TNFα and IL6 is significantly attenuated by overexpression of SOD1. El-Ansary et al 42 reported that lower concentrations of TNFα and IL6 in autistic patients may be related to overexpression of SOD, which was previously reported as metabolic biomarker in Saudi autistic patients. 43
Increasing evidence suggests that oxidative stress plays a role in the development and clinical manifestation of autism.44,45 In fact, oxidative stress plays a vital role in the pathology of several neurological diseases including Alzheimer disease, 46 Parkinson's disease, 47 Down syndrome, 48 schizophrenia, 49 major depressive disorder, 50 anxiety disorders such as panic disorder, 51 and obsessive-compulsive disorder. 52 It has been suggested that autism may result from an interaction between genetic, environmental, behavioral, and immunological factors, with oxidative stress as a mechanism linking these risk factors. No single gene has been found to be associated with autism, and involvement of multiple genes has been postulated.
Oxidative Stress and Autism
Oxidative stress occurs as a result of an imbalance between the production of reactive oxygen species (ROS) and endogenous antioxidants in living organisms. 53 Oxidative stress has been implicated in the pathogenesis of major psychiatric disorders, as the brain has comparatively greater vulnerability to oxidative damage than other organs. 54 ROS attack polyunsaturated fatty acids that are constitutive of cellular membranes, resulting in formation of lipid peroxidation (LPO) end products. Several reports have indicated increased levels of other LPO markers in autism, confirming an increase in oxidative stress in autism. Chauhan et al, 55 James et al, 56 and Al-Gadani et al 57 reported higher LPO in the blood of children with autism as compared to their developmentally normal, non-autistic siblings. LPO occurs as a chain reaction between polyunsaturated fatty acids and ROS, leading to the generation of lipid peroxides and hydrocarbon polymers, which are both highly toxic to the cell. Malonyldialdehyde (MDA) is generated by the peroxidation of polyunsaturated fatty acids and related esters and is used as a marker of LPO. 58 One study showed that plasma MDA content was higher in 13 of 15 (87%) of autistic subjects compared to in normal subjects. Zoroglu et al 59 reported increased thiobarbituric acid reactive species levels in the serum of autistic patients. In another study, the density of lipofuscin, a matrix of oxidized lipid and cross-linked protein that is formed as a result of oxidative injury in tissues, was observed to be higher in cortical brain areas concerned with social behavior and communication in autism. 60 Ming et al 61 reported increased excretion of 8-isoprostane F2alpha as a marker of oxidative stress in the urine of children with autism. Isoprostanes are produced from the free radical oxidation of arachidonic acid through non-enzymatic oxidation of cell membrane lipids.
It is also evident that increased oxidative stress in autistic children leads to decreased levels of non-enzymatic antioxidants such as glutathione (GSH), vitamin E, and ascorbic acid and disturb their metabolism, reducing the ability to fight oxidative stress. Antioxidant levels in patients with autism showed decreased activity of glutathione peroxidise (GPx) in plasma and in erythrocytes, 62 reduced levels of total GSH, a lower redox ratio of reduced GSH to oxidized glutathione (GSSG) in plasma,63–66 as well as decreased catalase and superoxide dismutase (SOD) activity in erythrocytes.59,62
Several studies have identified alterations in enzymes that play a vital role in the defense mechanism against ROS damage in autism. Yorbik et al 62 and Sogut et al 67 reported low activity of plasma antioxidant enzymes, particularly GPx and superoxide dismutase (SOD), in autistic children. Chauhan et al 55 reported higher MDA levels, while ceruloplasmin and transferrin levels were lower in autistic children. Increased levels of LPO together with decreased levels of serum ceruloplasmin and transferrin suggest that children with autism are under increased oxidative stress, likely due to abnormal metabolism of pro-oxidant metal ions and/or decreased antioxidant proteins. Evans et al 68 evaluated the oxidative stress metabolites carboxyethylpyrrole and iso[4]levuglandin (iso[4]LG) E2-protein adducts in cortical brain tissues in subjects diagnosed with autism. Significant immune reactivity towards all of these markers of oxidative damage in the white matter, which often extended well into the grey matter of axons, was observed in every case of autism examined. These investigators reported that a striking thread-like pattern appears to be a hallmark of the autistic brain, which was not observed in any age-matched control brain. In another study, the density of lipofuscin, a matrix of oxidized lipid and cross-linked protein that forms as a result of oxidative injury to the tissues, was observed to be higher in cortical brain areas concerned with communication in subjects diagnosed with autism. 69 Geier et al 66 reported well-defined increased levels of urine lipid peroxides and decreased levels of blood GSH and GPx in autistic patients. Similarly, Mostafa et al 70 demarcated that autistic children have increased oxidative stress resulting from enhanced LPO and/or decreased GPx by inducing autoimmunity in autistic patients, indicating the potential role of oxidative stress. Elevated levels of oxidative stress indicators such as NO, MDA, and protein carbonyl and reduced levels of antioxidant proteins such as ceruloplasmin and transferrin have also been reported in autistic children from the Sultanate of Oman.58,71
Neuroligins belong to a family of postsynaptic cell adhesion proteins that can bind to presynaptic proteins known asneurexins.72,73 Neuroligins are required for the maturation, stability, and/or maintenance of normal synaptic function.
74
Although four neuroligin genes have been identified in mammals, mutations in human genes encoding neuroligin 3 and neuroligin 4 have been recently associated with ASD.75,76 As a result, a new model for ASD has been developed that incorporates genetic-related changes to impaired synaptic processes in the pathobiology of autism.77,78 Evidence to support this model stems from a recent study showing that the absence of neuroligin-3 can induce nonsyndromic autism in mice.
79
A recent study showed that neuroligin-deficient mutants of
Mitochondrial Dysfunction and Impaired Energy Metabolism in Autism
Possible links between mitochondrial abnormalities and autism were initially suggested due to the deleterious consequences of mitochondrial disorders on neurodevelopment. Indeed, mitochondrial disorders often result in central nervous system (CNS) dysfunction, leading to developmental regression, learning disability, and various behavioral disturbances. Genetic mutations in mitochondrial DNA (mtDNA) have been associated with myopathy, cardiomyopathy, neuropathy, seizures, optic atrophy, strokes, hearing loss, diabetes mellitus, and other clinical features. 81 In some cases, autism can directly stem from mutations in mitochondrial DNA (mtDNA), as documented by Graf et al, 82 Fillano et al, 83 Pons et al, 84 Weissman et al, 85 Shoffner et al, 86 and álvarez-Iglesias et al. 87 The hypothesis of disturbed bioenergetics metabolism underlying autism has been suggested based on the detection of high lactate levels in some patients,88–90 and by nuclear magnetic resonance (NMR) imaging as well as positron emission tomography (PET) scanning, which documented abnormalities in brain metabolism. 91 A number of studies, including the detection of brain metabolism abnormalities and hyperlactacidemia, demonstrated a disturbance in brain energy metabolism in autistic patients, which may be a result of mitochondrial oxidative phosphorylation dysfunction in neuronal cells.82,84,91,92 Several mitochondrial respiratory chain disorders have been associated with autism.84,89,93 Hyperlactacidemia and an increased lactate/pyruvate ratio may result from several inherited metabolic defects of gluconeogenesis, pyruvate oxidation, the Krebs cycle, or the respiratory chain. Mutations in multiple genes may influence the observed changes in lactate and pyruvate levels, with genetic heterogeneity underlying mitochondrial dysfunction associated with autism.94–96
The higher levels of plasma pyruvate found in children with autism are consistent with the higher levels of alanine (transamination product of pyruvate) and lactic acid.97,98 Plasma pyruvate and lactate-to-pyruvate ratios suggest the presence of a pyruvate dehydrogenase complex (PDHC) deficiency, as previously reported.58,99,100 PDHC deficiency leads to in adequate removal of pyruvate and lactate, resulting in insufficient energy production. Hence, PDHC deficiency may contribute to brain dysfunction in autism. PDHC deficiencies are ascribed to oxidative modifications. Oxidative modifications and consequent inhibition of PDHC activity is consistent with the increased rate of hydrogen peroxide formation and reduced complex V activity observed in children with autism given the increased vulnerability of this complex to oxidative and nitrative stress.99,101 The lactate-to-pyruvate ratio accurately represents the redox state in the cytosol. For example, a lactate-to-pyruvate ratio of 12 in non-neurological controls is equivalent to a 750:1 ratio of oxidized nicotinamide adenine dinucleotide (NADH) to reduced NADH. Similarly, a lactate-to-pyruvate ratio of 6 reported in autistic children is equivalent to a 1500:1 ratio of oxidized NADH to reduced NADH. This more oxidized cytosolic redox state in autism may favor anaerobic glycolysis over oxidative phosphorylation as a source of adenosine-5′-triphosphate (ATP). Although skeletal muscle can tolerate this metabolic shift, consequences for brain function could be devastating due to the heavy reliance on mitochondrial oxidative phosphorylation for generating energy needed for cellular processes.100,102,103
The main function of the mammalian brain is generation and transmission of neural impulses. This requires creation and maintenance of ionic disequilibria, which, in terms of energy consumption, is a very costly process. In adult humans, the brain constitutes approximately 2% of body weight but receives 20% of the total blood supply. The brain utilizes oxygen at a rate of approximately 1.5 mmol/minute/g of tissue. Since the consumption of each mole of oxygen generates 6 mol of ATP during oxidative phosphorylation in the adult brain, at rest, 40%–60% of this high rate of ATP production and more during increased activity is used for ion movement. 104 The end products of brain energy metabolism, such as acetylcholine, ATP, and guanosine-triphosphate (GTP) are essential for powering basic cellular and molecular processes. Acetylcholine mediates learning, memory, and cognition, 105 while ATP and/or GTP are necessary for protein synthesis, 106 protein degradation, 107 synaptic transmission, 108 ion homeostasis, 109 and signal transduction. 110 Glutamate exposure reduces the oxygen consumption rate and ATP concentration in rat cerebral cortex neurons and has been implicated as a contributing factor in the pathogenesis of autism. 111 Disturbing energy metabolism makes brain cells more vulnerable to oxidative stress, glutamate-mediated neurotoxicity, and anoxic damage. 112
Mitochondria are membrane-enclosed organelles found in most eukaryotic cells, where they generate most of the cellular supply of ATP, which is used as a source of chemical energy. Additionally, mitochondria are involved in a range of other processes, such as signaling, cellular differentiation, cell death, and control of the cell cycle and cell growth. Mitochondrial dysfunction has been implicated in several neuropsychiatric disorders, particularly in depression, anxiety, schizophrenia, autism, and Alzheimer's dementia. Furthermore, the presence of mutations at mitochondrial or nuclear DNA (mtDNA and nDNA, respectively) levels has been linked to personality disorders, behavioral disturbances, thought alterations, impulsivity, learning impairment, cognitive impairment, and dementia. Mitochondrial dysfunction in the CNS of autistic individuals is supported by neuroimaging studies using PET and NMR spectroscopy, which have shown reduced glucose utilization and diminished ATP levels, particularly in the cerebral cortex.113,114 At the biochemical level, moderate lactic acidosis has frequently been observed in autism.88,95,98,115 Childhood mitochondrial respiratory chain disease involves impaired oxidative phosphorylation (OXPHOS) early in life, leading to ATP depletion and abnormal neuropsychological development.84,116 ASD is a well-recognized feature of childhood mitochondrial respiratory chain disease. Many patients with OXPHOS disorders possess a mutation within the mtDNA, which codes for 13 essential polypeptide components of the respiratory chain. Since mtDNA is nearly exclusively inherited from the mother, it is interesting that maternal factors also appear to be important in ASD, raising the possibility that mtDNA is a risk factor for developing the disorder.
Energy metabolism is central to life because cells cannot exist without an adequate supply of ATP. As previously mentioned, energy production for maintaining ionic disequilibria that is necessary for generation and transmission of nerve impulses is a primary function of the brain. The CNS is particularly sensitive to disturbances in energy generation; even a short-term interruption can lead to long-lasting, irreversible damage. If such a traumatic event occurs during birth, the consequences may last for a lifetime. Hence, understanding the relationship between oxidative stress, energy metabolism, and function during brain development has practical implications in humans. 117
The primary function of mitochondria is to produce ATP, the primary energy source in the brain and in the body. ATP, NADH, and nicotinamide adenine dinucleotide phosphate (NADPH) are important cofactors for many metabolic processes in the body. Many children with autism have lower muscle tone and decreased endurance; these symptoms may be related to decreased ATP levels.118,119 NADH is mainly involved in catabolic reactions (energy metabolism and mitochondrial function), whereas NADPH is involved in anabolic reactions (antioxidation and reductive biosynthesis). 120 Decreased plasma ATP may be related to impaired mitochondrial function, which has been reported in children with autism.85,95,96,121 Children with autism show significantly elevated oxidative stress, as indicated by the increased GSSG/GSH ratio (glutathione is the primary antioxidant in the body) and increased plasma nitrotyrosine. GSSG is reduced to GSH by glutathione reductase, and NADPH. NADPH levels were substantially lower in children with autism. Low levels of plasma ATP reduced NADPH and plasma tryptophan. 122 Although glutathione reductase and glucose-6-phosphate dehydrogenase are secondary antioxidant enzymes that help maintain a steady concentration of glutathione, NADPH is necessary for optimal functioning of the primary antioxidant enzymes.123,124 This may be due to impaired function and/or amount of trans locator protein in the mitochondrial membrane, which would impair transport of ATP from the mitochondria into the cytoplasm and into the plasma.
Superoxide and hydroxyl radicals are ROS that can rapidly overwhelm endogenous scavenging mechanisms, damaging cellular macromolecules, including lipids, proteins, and nucleic acids. Oxidants are also mediators in signaling involving mitochondria, DNA repair enzymes, and transcription factors that may lead to apoptosis during reperfusion. Mitochondria are heavily implicated in the formation of ROS since excessive superoxide production during electron transport and inhibition of electron transport mechanisms by free radicals leads to further generation of oxygen radicals. 125 Mitochondrial oxygen radical production can be stimulated by impaired intracellular calcium, sodium, and adenosine diphosphate.122,126,127 Multiple lines of evidence suggest that the influence of adenosine may be insufficient for causing ASD, and that increased adenosine would reduce both physiological and behavioral hallmarks of ASD. 128
The multi-subunit NADH-ubiquinone oxidoreductase (complex I) is the first enzyme complex in the mitochondrial electron transport chain (ETC). The iron–sulfur protein fraction of complex 1 is made up of seven subunits, including NADH-ubiquinone oxidoreductase 1 alpha sub complex 5 (NDUFA5). The NDUFA5 gene is located on human chromosome7q32, which is a candidate region for autism. 129 Marui et al 130 demonstrated an association between the NDUFA5 gene and autism. The NDUFA5 gene product is part of an enzyme complex involved in the mitochondrial ETC. Variations in this gene and mitochondrial dysfunction may play important roles in the etiology of autism.
Heguilen et al 131 and Lina et al 132 reported that Na+/K+-ATPase and Ca2+/Mg2+-ATPase activities in autism may be increased in response to increased intracellular calcium concentration, which may contribute to altered neocortical circuitry in the cerebellum and frontal cortex of individuals with autism. Al-Mosalem et al 133 observed lower ATP levels in red blood cells as well as elevated lactate and creatinine kinase (CK) activities in the plasma of Saudi autistic children compared to age-matched controls. Furthermore, activation of brain CK may be related to significant increases in the activities of Na+/K+ATPase and Ca2+/Mg2+ ATPase and marked decreases in expression of mitochondrial ETC complexes in different regions of the brain in autistic subjects compared with their age-matched controls. This suggests that these enzymes contribute to abnormal energy circuit functioning in autism.134,135 Creatine is a nitrogencontaining compound that serves as an energy shuttle between mitochondrial sites of ATP production and the cytosol, where ATP is utilized. Disorders of creatine synthesis, characterized by brain creatinine deficiency, can lead to numerous deleterious symptoms including mental retardation, hypotonia, autism or behavioral problems, and seizures. Diagnosis of these conditions relies on plasma and urine creatine levels. Creatine levels in plasma are reduced in both creatine synthesis defects. The urine creatine/creatinine ratio is elevated in creatine transporter deficiency with normal plasma levels of creatine. The diagnosis is confirmed in all cases through DNA testing or functional studies. 136
Mitochondria are responsible for most of the energy production through oxidative phosphorylation, a process requiring the action of various respiratory enzyme complexes, and the mitochondrial ETC, which is located in the inner mitochondrial membrane. 137 Mitochondria produce ATP by generating a proton gradient (membrane potential) through the action of five ETC complexes, including complex 1 (NADH dehydrogenase), complex 2 (succinate dehydrogenase), complex 3 (cytochrome bc1 complex), complex 4 (cytochrome c oxidase), and ATP synthase, also known as complex 5. Here, electron transport couples with the translocation of protons from the mitochondrial matrix to the inter membrane space. The generated proton gradient is used by ATP synthase to catalyze the formation of ATP by phosphorylating ADP. 138 The number of mitochondria per cell is roughly related to the energy demands of the cell. The brain has a high energy demand; thus, neurons contain a large number of mitochondria. The ETC in mitochondria is also a primary mechanism for free radical generation.139,140 Changes in the mitochondrial ETC have been suggested as contributing factors in the pathogenesis of several diseases, including neuropsychiatric and neurodegenerative disorders.141–143 Chauhan et al 135 showed that the expression of ETC complexes (1, 2, 3, 4, and 5) is decreased in the cerebellum as well as frontal and temporal regions of the brain in children with autism, leading to abnormal energy metabolism and increased vulnerability to oxidative stress. Autism is associated with a deficiency in complexes 3 and 4 of the mitochondrial respiratory chain, leading to an elevated lactate-to-pyruvate ratio.85,144,145
Adiponectin is a protein that is produced by adipose tissue and involved in the control of energy metabolism.
146
Previous studies in Japan, Italy, and the Sultanate of Oman have suggested that altered levels of adiponectin reflect impairments in social interaction, and may be implicated in the pathophysiology of autism.147–149 Leptin, a peptide hormone mainly secreted by adipose tissue, is involved in the regulation of body weight and energy expenditure at the hypothalamic level. Significantly higher levels of leptin were found in Omani autistic children. This may be involved in the regulation of neuroendocrine functions, the immune system, and inflammatory response, as well as play a role in development. Involvement of some cytokine-like hormones and proinflammatory adipokines have been implicated in the pathophysiology of autism.
150
Potential mechanism of oxidative stress in autism proposed by Chauhan and Chauhan, (2006).
Tryptophan Metabolism in Autism
L-tryptophan is an essential amino acid that cannot be synthesized in the human body and must be supplied by the diet. For all amino acids, including L-tryptophan, only the L isomer is used in protein synthesis and can cross the blood–brain barrier. In humans, tissue storage of tryptophan is relatively low and the overall tryptophan concentration in the body is the lowest among all amino acids, although only small amounts are necessary for general healthy nutrition. Common sources of tryptophan include oats, bananas, dried prunes, milk, tuna fish, cheese, bread, chicken, turkey, peanuts, chocolate, and fruits such as cherries.151–153 The kynurenine pathway (KP) is a major route of L-tryptophan catabolism, resulting in the production of the important pyridine nucleotide nicotinamide adenine dinucleotide (NAD+) and several neuroactive intermediates. Tryptophan plays an important role in protein biosynthesis and as a precursor of various metabolic pathways (Fig. 2), including serotonin, melatonin, kynurenine, and quinolinic acid (QUIN).
153
Its metabolism leads to production of many metabolites that may be physiologically valuable or even toxic.
154
Catabolic pathway of tryptophan.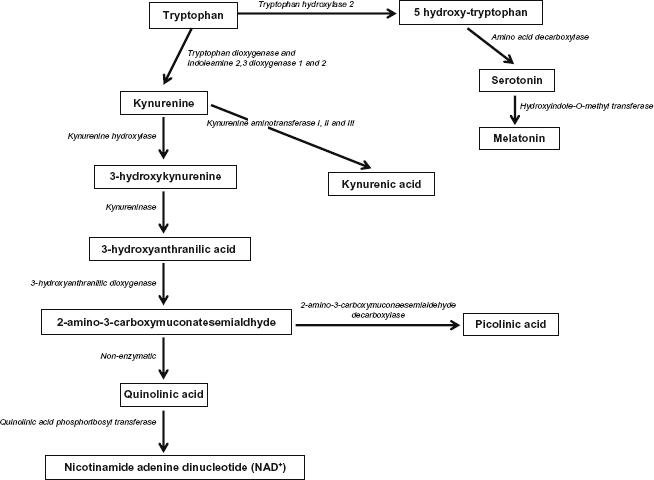
Current estimates suggest that 95% of mammalian serotonin is localized within the gastrointestinal tract, and only 3% of dietary tryptophan is utilized for serotonin synthesis in the body. However, serotonin synthesis is one of the most important tryptophan pathways. Only 1% of dietary tryptophan is used for serotonin synthesis in the brain. Despite the relatively low concentration of brain serotonin compared to that in the body, it has a broad impact as a neurotransmitter and neuromodulator and has been implicated in numerous psychiatric conditions and psychological processes, including autism.90,122 D'Eufemia 155 suggested that low brain tryptophan availability due to a low serum Try/LNAA (large natural amino acids) ratio may be one of the mechanisms involved in altering serotonergic function in autism, as well as lower total plasma tryptophan and Try/LNAA ratio in people with signs of behavioral disturbances and depression. Tryptophan is a precursor to the neurotransmitter serotonin, which is strongly linked to autism. Lower levels of tryptophan may play a role in pediatric disorders. Evans et al 68 and Kałuzna-Czaplinska et al 156 suggested that urinary levels of tryptophan are reduced in autistic children. This may lead to worsening of autistic symptoms such as depression and increased irritability. Serotonin deficiency is linked to the pathophysiology of many psychiatric disorders, ranging from depression, anxiety, obsessive-compulsive disorder, eating disorders, and dependence. 157 Psychiatric complications of neurodegeneration may originate from serotonin deficiency mediated by tryptophan depletion following KP activation.
Monoamine oxidase exists in two isoforms (MAO A and MAO B). MAO A plays a vital role in metabolizing neurotransmitters such as dopamine, norepinephrine, epinephrine, and serotonin. Abnormalities in serotonin neurotransmission have long been implicated in the psychopathology of autism. Dysfunctions of MAO A have been implicated in a variety of neuropsychiatric disorders, including depression, social anxiety, autism, and attention deficit hyperactivity disorder. Essa et al 158 reported reduced levels of MAO A activity in children with autism, which may affect the metabolism of neurotransmitters associated with autism pathogenesis.
Melatonin is an indoleamine synthesized from tryptophan in the pineal gland by pinealocytes. Physiologically, melatonin functions to regulate the sleep–wake cycle. It is also an important antioxidant and scavenger of free radicals. Additionally, melatonin has significant immune regulatory functions. Reported beneficial effects of melatonin in ASD include improved sleep, reduced “sun downing,” and reduced cognitive decline. Nonetheless, inflammation associated with neurodegeneration may lead to melatonin deficiency through KP and through the mediation of tryptophan depletion. This deficiency would be expected to compromise melatonin-mediated antioxidant and free radical scavenger defenses, both of which are important in the pathogenesis of neurodegeneration. 159
Tryptophan depletion may represent an immune-mediated mechanism involved in the pathophysiology of autism, or it may exacerbate autistic symptoms during immunological stress. Immune dysregulation during brain development can have dramatic effects on neuronal function.160–162 Increasing evidence suggests that immune dysregulation is associated with neurodevelopmental disorders, including ASD.163,164 Induction of the KP is associated with significant immunomodulatory changes, for which two non-mutually exclusive mechanisms have been proposed. First, KP activation results in tryptophan depletion and immune response impairment by reducing the availability of this essential amino acid. Second, actions of downstream metabolites of the KP suppress the immune system. For example, increased KP activity in dendritic cells is associated with the complete blockade of clonal T-cell expansion; tryptophan depletion and KP activation have been implicated in the development of immune tolerance associated with pregnancy and persistence of tumours. 159
QUIN is an important excitotoxic metabolite of the KP; there is accumulating evidence that QUIN is involved in neurotoxicity associated with several inflammatory brain diseases. Like glutamate, QUIN is an agonist of the N-methyl-D-aspartate receptor and can induce excitotoxicity in human neurons and astrocytes.165,166 Our group recently showed that QUIN can induce poly (ADP-ribose) polymerase (PARP) activation and subsequent intracellular NAD+ depletion and ATP stores, leading to a number of deleterious effects, such as mitochondrial permeability, overproduction of superoxide, and nitric oxide (NO) production, which are relevant to the pathogenesis of autism and ASD. 166
Interestingly, Zimmerman et al 167 reported a decrease in QUIN levels in the cerebrospinal fluid from autistic children. This suggests that inflammation is impaired in the autistic brain. We previously demonstrated that fetal brain cells can only produce small amounts of QUIN due to lower enzyme capacity. Therefore, the KP may not be fully functional in the CNS of autistic individuals. Moreover, QUIN formation from tryptophan may be impaired. The enzyme indoleamine 2,3-dioxygenase is the primary enzyme in the KP. Analogous to this is tryptophan 2,3-dioxygenase, for which a genetic polymorphism in autism has already been described. 168 The KP is associated with the formation of serotonin since it is generated from tryptophan through the catalytic activity of the enzyme tryptophan 5-hydroxylase (THP). Most importantly, a THP2 gene polymorphism is clearly associated with autism and other neurological disorders. 169 Therefore, impaired synthesis of both QUIN and serotonin in autism may be related through dysregulated KP metabolism, which is likely due to abnormal mRNA and protein expression of KP enzymes, leading to altered cell signaling during early development.
NAD+ is an end product of tryptophan catabolism. The importance of maintaining intracellular NAD+ and NADH levels for maintaining overall cellular integrity and function is well established. Reduced NAD+ levels represent a potential pathogenic mechanism for cell death. Additionally, NAD+ is an important contributor to energy (ATP) production and serves as a cofactor for NAD glycohydrolases involved in intracellular calcium regulation. Furthermore, NAD+ is cofactor for the DNA repair enzyme PARP, an essential intracellular enzyme for repair of DNA damage caused by increased ROS levels. Excessive PARP activity results in depletion of intracellular NAD+ levels and contributes to cell death via energy restriction. Thus, continuous biosynthesis of NAD+ is vital for maintaining cell viability. Increased levels of NADH with decreased levels of ATP, NAD+, and the NAD/NADH ratio, and plasma tryptophan occurring in parallel with increased levels of oxidative stress has been observed in children with autism.45,58,122,170 Therefore, we propose that impairing tryptophan has a major impact on the neurotoxic cascade observed in autism.
Conclusion
This review indicates that understanding the role of oxidative stress may provide renewed insight into the pathophysiology of autism, which is likely affected by the environment, genetic factors, immunological factors, new treatments, and prevention. Increased oxidative stress in autism may be due to (a) increased production of oxidative markers or (b) increased exposure to environmental pro-oxidants, or (c) deficiencies of antioxidants (ceruloplasmin, transferrin, SOD, GPx, catalase, reduced glutathione), impaired energy metabolism (NAD+, NADH, ATP, pyruvate, and lactate), and abnormal tryptophan catabolism. Increased oxidative stress may lead to membrane lipid abnormalities, mitochondrial dysfunction, excitotoxicity, inflammation, and immune dysregulation in autism. These abnormalities may contribute to behavioral abnormalities, sleep–wake disorder, and gastrointestinal disturbances reported in autism.
Author Contributions
Conceived and designed the experiments: MME, SS, NB, SA-A. Wrote the first draft of the manuscript: MME, SS, NB, SA-A, TM. Contributed to the writing of the manuscript: MME, SS, NB, CKL, SA-A, TM. Agree with manuscript results and conclusions: MME, NB, SA-A, GJG, TM. Jointly developed the structure and arguments for the paper: MME, NB. Made critical revisions and approved final version: MME, NB, GJG. All authors reviewed and approved of the final manuscript.
Funding
The project was supported by Sultan Qaboos University; Oman in the form of internal grant is gratefully acknowledged (IG/AGR/FOOD/11/02). This work has been also supported by the National Health and Medical Research Council (NHMRC) and by the Rebecca Cooper foundation (Australia). Nady Braidy is the recipient of the Alzheimer's Australia Viertel Foundation Postdoctoral Research Fellowship at the University of New South Wales. Postdoctoral fellowship offered to Subash Selvaraju from The research Council Oman (RC/AGR FOOD/11/01) is gratefully acknowledged.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Eethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.