
Abstract
Emergent seizures are common in Alzheimer's disease (AD), although the mechanisms mediating this are unknown. It is proposed that stress induced interleukin-18 (IL-18), via interferon-gamma (IFNy) and independently, increases indoleamine 2,3-dioxygenase (IDO) and subsequent quinolinic acid (QA) in microglia. QA increases seizures and concurrently contributes to neuronal loss via excitotoxicity. The ApoE4 allele interacts with IL-18 polymorphisms to increase the risk of AD, and seems likely to potentiate the emergence of seizures. Concurrent changes in IDO and the kynurenine pathways at the blood-brain-barrier (BBB) have implications for treatment, including in the efficacy of different anti-hypertensives. Melatonin is proposed to inhibit these overlapping excitotoxic and neurodegenerative processes, and would be a useful adjunctive treatment.
Alzheimer's Disease and Seizures
There is growing data showing an increased association of seizures with Alzheimer's, in both humans and animal models.1,2 Estimates of prevalence vary, but it seems that about 1.5% to 10% of people with Alzheimer's may experience seizure activity, with the highest prevalence in early onset Alzheimer's. 3 This raises the question as to whether there is a subtype of Alzheimer's that is seizure associated and may be linked to differential changes and possibly to differential treatment.
Quinolinic acid (QA) is a possible mediator of both seizures and neuronal loss.4,5 In the brain, microglia are the most likely source for QA. QA mediates neuronal excitotoxicity via the N-methyl-D-aspartate receptor (NMDAr) and is usually induced by interferon-gamma (IFNy), 6 although other factors are known to mediate an increase in the levels of indoleamine 2,3-dioxygenase (IDO), and subsequently QA.7,8 One such factor is IL-18.
IL-18 is induced by stress, 9 including in neurons. 10 It is cleaved within the cell by Caspase-1, like IL-1beta, and when released mediates an increase in IFNy. 11 Such Caspase-1 activation has upstream links to inflammasome induction, and therefore to wider models of neurodegeneration. 12 It is therefore possible that IL-18, including via IFNy, could be associated with an increase in the levels of IDO activity and QA induction in microglia. IL-18 has been recently shown to increase glycogen synthase kinase 3-beta (GSK-3b) and tau hyperphosphorylation. 13 Would variations in the levels of IL-18 be relevant to early onset seizure associated Alzheimer's?
IL-18 has been shown to be increased in the brain in Alzheimer's, and increased in the cerebral spinal fluid in mild cognitive impairment, 14 and IL-18 polymorphisms are associated with an increase in Alzheimer's susceptibility, showing synergistic interactions with the ApoE4 allele. 15 Interestingly the ApoE4 allele, independent of dementia, is associated with an increase in the susceptibility to seizures. 16 As to whether IL-18 polymorphisms or increases in IL-18 would synergistically interact with the ApoE4 allele to induce an increase in seizures as well as Alzheimer's remains to be examined. It would be expected that IL-18, via an increase in GSK-3b, would increase the hyperphosphorylation of tau and enhance Amyloid B (AB) production. 17 Recent data shows that AB may prime microglia-like cells for a sub-threshold concentration of IFNy to induce IDO/QA. 18 60% of IDO induction in AB primed cells is mediated by an IFNy induced increase in tumor necrosis factor alpha (TNFa), and the subsequent autocrine effects of TNFa. Previous data 19 in this cell line show that AB effects are prevented when the sphingosine-1-phosphate receptor 1 (S1P1r) is k.o.'d. Would variations in the levels/activity of the S1P1r be a significant modulator of such AB priming for subsequent IFNy? This awaits experimental data, but it would suggest that the effects of AB, like LPS or thrombin, in microglia is determined by an increase in the levels of GSK-3b and enhanced NADPH Oxidase activation. 20 This would then modulate the S1P/Ceramide ratio, as part of wider oxidant status driven lipid raft re-organization. 21 Presumably factors that increase the levels of endogenous anti-oxidants will modulate this oxidant driven priming and raft re-organization. A number of factors inhibit GSK-3b and NADPH Oxidase in microglia, including lithium, 22 resveratrol, 23 and melatonin. 24 All are associated with an increase in the phosphorylation, and inhibition, of GSK-3b, and therefore leading to an increase in NF-E2-related factor (Nrf-2) and endogenous anti-oxidants. Modulation of microglia reactivity threshold may be mediated by this.
How IL-18 induced IFNy impacts on AB primed microglia awaits further experiments. However, it is possible that IL-18, independent of IFNy, can increase IDO, as shown in other cell types. 25 Would IL-18 directly mediate an increase in IDO? Some unpublished data suggests that this could be so. 26 As to whether AB primes microglia for IL-18, as it does for IFNy, remains to be determined. IL-18 seems to play a role in changing the most relevant factors associated with Alzheimer's through its impact on tau, AB and possibly microglia threshold. IL-18 is currently being investigated for impacts at the blood brain barrier (BBB). Should it induce GSK-3b and alter oxidant status in the BBB, then this could overlap it to the effects of peripheral LPS, which mediates changes in RAGE (Receptor for Advanced Glycation End Products) and LRP-1 (low density lipoprotein receptor-related protein-1). 27 Such changes lead to an increase in the influx and decrease in the efflux of AB over the BBB. Would IL-18, perhaps in conjunction with AB, parallel such oxidant associated changes in the BBB?
In human endothelia and pericyte lines, LPS leads to an increase in the levels of abluminally released kynurenine. 28 This seems likely to be taken up by astrocytic end-feet and rapidly converted to kynurenic acid (KA). Such an increase in astrocyte KA is likely to mediate LPS induced depression. Would IL-18, with or without AB priming, modulate the IDO and kynurenine pathways at the BBB? Such putative increases in KA would likely be anti-epileptic, although it would also induce cognitive impairment, as in schizophrenia as well as in Alzheimer's. 29 This is presuming KA release from astrocytes, perhaps in a targeted manner, to neurons. However, in an inflammatory context, where BBB changes are occurring, then microglia may have closer proximity to the BBB, in conjunction with perivascular macrophages, 30 and the kynurenine produced could be driven to QA production, and therefore contribute to excitotoxicity, and seizure induction. An increase in kynurenine, QA and KA would suggest that more tryptophan is being driven down the kynurenine pathway, and less to serotonin and melatonin formation.
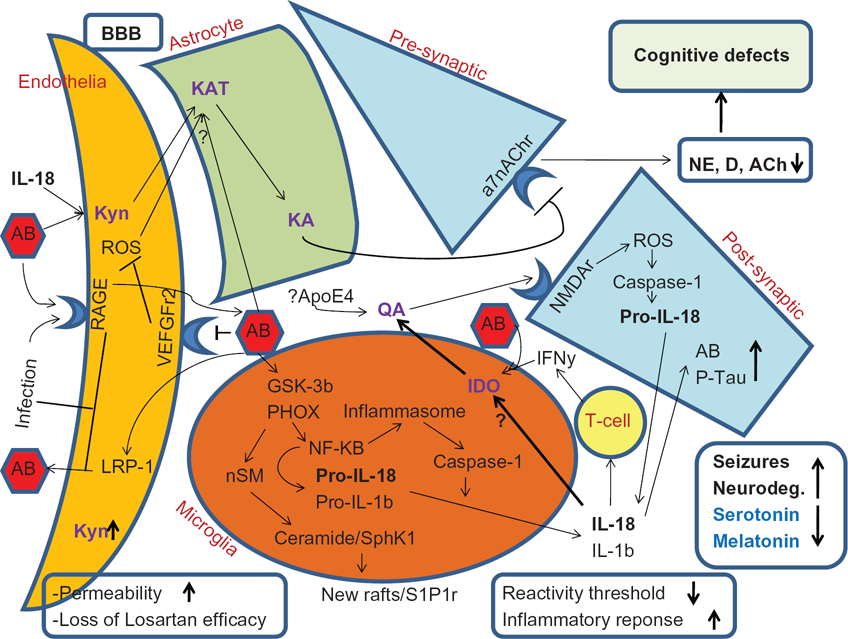
Showing the proposed pathways whereby stress and IL-18 polymorphisms mediate an increase in IL-18, with impacts concurrently on seizures neuroexcitotoxicity and neurodegeneration. Infection effects on RAGE activity and LRP-1 levels will lead to an increase in AB in the brain. Via the inhibition of the VEGFr2, then AB will inhibit the efficacy of Losartan/EXP3179, with concurrent changes in permeability. In microglia AB will lead to a decrease in the reactivity threshold, likely involving S1Pr activation, within the context of wider rearranged lipid raft complexes. AB will lower the threshold for IFNy induced IDO, perhaps in part via autocrine TNFa, leading to an increase in QA. IL-18 will be induced in both glia and neurons, via ROS induced inflammasome and Caspase-1 induction. Decreases in Dopamine, NE, and Ach will contribute to cognitive difficulties. The decrease in D and NE, via the loss of cAMP induction, may contribute to a decrease in astrocyte KA production. Such increases in IDO will drive tryptophan down the kynurenine pathway, leading to a decrease in the levels of serotonin and melatonin, further exacerbating mood and oxidant status. ApoE4 interacts with IL-18 polymorphisms in mediating an increase in sporadic Alzheimer's, and this interaction may increase the likelihood of concurrent seizures, either directly and/or indirectly via an increase in QA. Melatonin will have multiple sites of action, via changes in oxidant status in all cell types. Amyloid B and IL-18 effects are not shown in astrocytes for clarity.
Such changes at the BBB are likely to impact on the effects of medications. For example, Losartan, via its active metabolite EXP 3179, mediates an increase in the Akt/pGSK-3b/Nrf-2/anti-oxidant paths. This is achieved via the induction of vascular endothelial growth factor (VEGF) and its effects at the VEGF receptor 2 (VEGF R2). 31 AB directly blocks the VEGF R2, 32 and so some of the anti-hypertensive and importantly anti-oxidant effects of Losartan will be lost. A shift to other anti-hypertensives may be useful. This would highlight the need for more dynamic and reactive prescribing, which in turn would be dependent on a more detailed knowledge of the processes of change, including the changes that seizure induction may indicate.
Melatonin has also been shown to be protective in Alzheimer's, 33 and has been shown to significantly decrease the levels of GSK-3b. 34 Would a significant decrease in melatonin interact with seizure susceptibility in Alzheimer's? No current data directly answers this, although melatonin is known to have anti-epileptic effects. 35 Melatonin also reverses some of the other cellular changes that are associated with Alzheimer's, including a decrease in the longevity protein sirtuin-1, 36 a decrease in PGC-1a (peroxisome proliferator-activated receptor-gamma, coactivator-1alpha) 37 and a decrease in oxidative phosphorylation. 38 Would variations in melatonin be associated with seizure susceptibility, and perhaps the modulation of the IL-18/IFNy induced increases in IDO/QA in seizure associated Alzheimer's? Melatonin is known to modulate BBB permeability, and this may be relevant to changes in brain AB levels, as suggested above. Also the kynurenine aminotransferases (KATs) are sensitive to oxidative stress. 39 A decrease in KA arising from such oxidative modulation may allow more kynurenine to form QA. Melatonin would likely modulate this. There is one paper showing that astrocytes are able to produce melatonin when adequate serotonin is present. 40 Given the anti-seizure, anti-cortisol and cortisol modulating effects of melatonin, 41 then such induction by astrocytes would be a potential local target for drugs to induce. It would seem likely that such astrocyte derived melatonin would modulate seizures, stress/IL-18 as well as microglia and BBB oxidant status and associated changes, as shown in the Summary Figure.
In conclusion, an argument can be made for the role of stress induced increases in IL-18, perhaps via IFNy and/or AB priming, in the modulation of IDO in both microglia and endothelia. In microglia an increase in the levels of QA would be associated with both seizures and neuronal loss, and these effects may be potentiated by the ApoE4 allele. As to whether such pathways constitute a sub-type of Alzheimer's, or are differentially activated along a continuum in all people with Alzheimer's remains to be determined. Further research on the processes may lead to a better treatment strategy for this putative sub-type of Alzheimer's.
Disclosure
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.