
Abstract
The regions encoding the coordinately regulated Th2 cytokines IL5, IL4 and IL13 are located on chromosomes 5 of man and 11 of mouse. They have been intensively studied because these interleukins have protective roles in helminth infections, but may lead to detrimental effects such as allergy, asthma, and fibrosis in lung and liver. We added to previous studies by comparing sequences of syntenic regions on chromosome 3 of the rabbit (Oryctolagus cuniculus) genome OryCun 2.0 assembly from a tuberculosis-susceptible strain, with the corresponding region of ENCODE ENm002 from a normal rabbit as well as with 9 other mammalian species. We searched for rabbit transcription factor binding sites in putative promoter and other non-coding regions of IL5, RAD50, IL13 and IL4. Although we identified several differences between the two donor rabbits in coding and non-coding regions of potential functional significance, confirmation awaits additional sequencing of other rabbits.
Introduction
Rabbits (Oryctolagus cuniculus), a valuable resource for diagnostic and therapeutic antibodies, are becoming increasingly important for vaccine development. The unique characteristics of their immune system make them a major source of antibodies of high affinity and specificity. Rabbits have long been models for human infectious diseases and more recently for autoimmune, neurological, ophthalmological, respiratory and cardiovascular diseases. They are widely used in development of surgical techniques, testing of therapeutics, and are also valued as a source of fur and meat in many parts of the world.
Annotation and analysis of the rabbit genome is therefore of importance for both biomedicine and agriculture and is of special importance to immunologists. NCBI maintains a Rabbit Genome Resources website (http://www.ncbi.nlm.nih.gov/projects/genome/guide/rabbit/).
The Broad Institute has submitted the second whole genome assembly of the European rabbit, completed at 6.51x coverage, to GenBank. The assembly is available in MapViewer as OryCun, build 2.0. The NIH Intramural Sequencing Center (NISC) performed clone-based sequencing of regions of the rabbit genome as part of the NISC ENCyclopedia Of DNA Elements (ENCODE) comparative sequencing project 1 and deposited the sequences in GenBank.
The ENCODE project and the Broad Institute sequenced rabbits with different genealogies and phenotypes. ENCODE sequenced an outbred New Zealand White (NZW) rabbit, whereas the Broad Institute sequenced a rabbit of the partially inbred “Thorbecke” NZW strain. The Thorbecke rabbit may have had significant immunological, physiological, and developmental abnormalities. Dorman et al 2 report that the phenotype included “ruffled fur, narrow palpebral fissures and stunted facies” and furthermore “abnormal closeness of eyes, lop ears in some animals and sedentary behavior”. Rabbits of the Thorbecke strain had greater susceptibility to M. tuberculosis infection. 2 Despite the phenotypic abnormalities, the Thorbecke strain was chosen for sequencing at Broad Institute because it was less heterozygous than outbred NZW (personal communication to RGM). Regrettably, all Thorbecke rabbits were lost in a fire in January 2005.
In both assemblies of rabbit, the cytokine genes Interleukin 4 (IL4), Interleukin 13 (IL13), and Interleukin 5 (IL5) were placed near each other in the “Th2 cytokine region”, with synteny to corresponding regions in human and mouse. The Broad Institute assigned the region to rabbit chromosome 3. The Th2 region, and the IL4 cytokine in particular, have been linked to the progression and severity of tuberculosis.3–5 It was of interest to learn whether any variants in the region with IL5, IL4, IL13, and other nearby genes (RAD50, KIF3A) could have contributed to immune system deficits in the Thorbecke rabbit.
The cytokines encoded in the Th2 region are characteristic of type 2 immunity. Type 2 immunity has important protective roles in responses to helminth infections, but detrimental effects include allergy-associated IL4-induced elevations in serum IgE, IL5-induced eosinophilia and airway remodeling in asthma, and IL13-induced epithelial cell damage leading to fibrosis in lung, or in liver, during helminth infections. 6 The Th2 region was selected for sequencing by ENCODE because of the important roles that cytokines play in determining the developmental fate and effector functions of T lymphocytes in the immune system. 7 The expression of IL4, IL13 and IL5 in this region is coordinately regulated, and the finding of conserved non-coding regions suggests that the mechanism of regulation is also conserved in syntenic regions of other species. 8 The conserved structure of the Th2 region is shown in Figure 1.
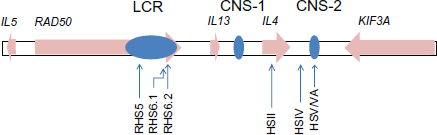
Conserved structure of the Th2 region. A schematic (not drawn to scale), of the structure of the Th2 region, conserved across many species, including those used in this study. The genes in the region are IL5, RAD50, IL13, IL4, and KIF3A, and the direction of transcription is shown using arrows. The locus control region, near the end of the RAD50 gene, is labeled LCR.
To identify conserved noncoding sequences in the Th2 region, we conducted comparative genome sequence analysis in 10 mammalian species including the rabbit, mouse, and human. Previous studies have used a functional approach, usually in mice, to define roles for various transcription factors in the Th2 cytokine region. Among the transcription factors known to bind to at least one location in the region are Ets-1, 9 GATA3,10,11 c-Maf, 12 RBPJK, 13 Runx3, 14 IRF4, 15 JunB 16 and STAT family members.17,18 Strempel et al 9 did multi-species bioinformatic comparisons to reach predictions of only Ets-1 and GATA binding sites, but their work included neither other transcription factors nor the rabbit.
We sought to address three general questions:
Do the Broad and ENCODE assemblies of the Th2 region differ in gene content, and is it possible that these differences had phenotypic consequences?
Are the sites predicted by Strempel et al 9 conserved in rabbit, and if so, what are the rabbit-specific binding sites?
Can we find transcription factor binding sites (TFBS) conserved across mammals for some transcription factors other than Ets-1 and GATA?
Results
Genomic sequences
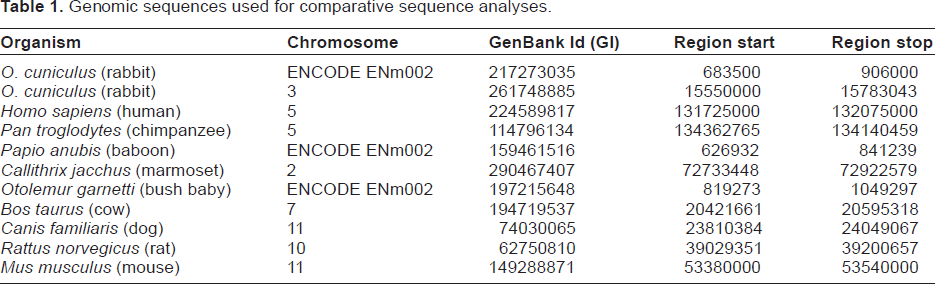
We studied genomic sequences containing the genes IL5, RAD50, IL13, IL4 and KIF3A from rabbit and the nine species used by Strempel et al. 9 Table 1 lists the species and the genomic sequences used in this study.
Genomic sequences used for comparative sequence analyses.
We considered the possibility of adding additional species to the study. A Th2 region syntenic to that in human exists in chicken (Gallus gallus). 19 We did not use the chicken genome because we found few conserved non-coding regions in chicken by a Mulan alignment (data not shown). Strempel et al 9 state the same reason for not using chicken. As of March 2011, the only other whole genome sequence in NCBI MapViewer that has a clear Th2 region belongs to Sumatran orangutan (Pongo abelii), which we did not add to the study since we already include two species of great ape, human and chimpanzee.
Comparison of the Broad and Encode Sequences within Predicted genes
We compared the Broad and ENCODE sequences and annotations of the genes IL5, RAD50, IL4, IL13, and KIF3A. We were able to confirm, by alignment, the placement of most exons in these genes (see Supplementary Data). The exceptions were that exons 4 and 5 of IL5 could not be placed on the ENCODE sequence, that exon 6 of RAD50 could not be placed on the Broad sequence, and that the ENCODE annotation did not include what Broad annotates as exons 10 and 11 of KIF3A. Further analysis suggests that RAD50 was misassembled in Broad and that there exists insufficient evidence to support Broad's prediction of putative exons 10 and 11 in KIF3A.
We compared the assembled coding regions of these five genes (see Supplementary Data for details). We found a substitution of a Threonine (Thr) in Broad for a Proline (Pro) in ENCODE at amino acid 27 of IL13. The substitution is supported by traces in the NCBI trace archive. In-silico structural analysis and comparison with homologous sequences suggest that both Thr27 and Pro27 would be tolerated.
Of possible immunological interest, there is a frameshift mutation in exon 2 of IL4 in the Broad assembly. This frameshift is supported by the trace with identifier 2047213760. A second trace, identifier 2061258363, aligns with the single nucleotide insertion, but has two gaps elsewhere in the alignment. Because the coverage of this position in IL4 is at most 2x, the evidence for the insertion is weak. No traces matched the ENCODE/wild-type sequence, so there is no evidence that the sequenced rabbit was heterozygous for the IL4 single-nucleotide insertion.
See Figure 2 for alignments of the rabbit, human and mouse protein sequences of the genes IL5, IL13, and IL4.

Alignments of the rabbit, human, and mouse protein sequences for IL5, IL13, and IL4. Panel
Comparison of the Broad and Encode Promoter sequences
We aligned promoter sequences for IL5, RAD50, IL13, and IL4 from the ENCODE genomic sequences to the Broad assembly; see Supplementary Data. The ENCODE RAD50 and IL13 promoter sequences align to the Broad assembly with full coverage and high percent identity. The Broad IL5 and IL4 promoters matched the ENCODE sequences well, but the Broad sequences had runs of the ambiguity character N that split the alignment into partial matches. Because the promoter regions in ENCODE do not contain Ns, we used the ENCODE sequences for cross-species comparison and de-novo prediction of binding sites.
Placement of Ets-1 and Gata Binding sites
We placed the Ets-1 and GATA binding sites described in Strempel et al 9 on both rabbit assemblies using two methods. The first method was direct alignment by BLAST 20 of the sequences provided by Strempel et al. 9 The second method was to use the Mulan 21 and multiTF algorithms to place the binding sites. These placement methods gave similar results, but they differ from the results of Strempel et al 9 in part because Strempel et al 9 used the MatInspector program, rather than multiTF, to predict binding sites. MatInspector uses a proprietary library, and we cannot use the program due to the restrictive license on how annotations generated by MatInspector may be published.
Ets-1 and Gata Binding Sites Placed Using BLAST
Twelve of the 19 Ets-1 and GATA transcription binding sites could be unambiguously placed on both the Broad and ENCODE assemblies by alignment to the homologous sequences in the other nine species. The locations of these binding sites are shown in Table 2.
Ets-1 and GATA binding sites that could be placed by BLAST.
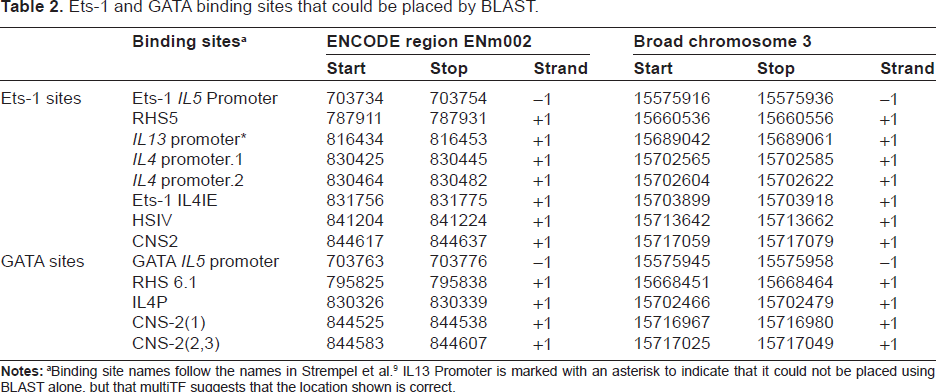
Binding site names follow the names in Strempel et al. 9 IL13 Promoter is marked with an asterisk to indicate that it could not be placed using BLAST alone, but that multiTF suggests that the location shown is correct.
Each site in Table 2 aligns to the homologous sequence of at least eight of the species with coverage of at least 80% and E-value of at most 0.1, except HSIV and Ets-1 IL13 Promoter. Ets-1 IL13 Promoter cannot be confidently placed by BLAST alone, as only three homologous sequences aligned to rabbit regions with the required coverage and E-value cutoff. The multiTF program, however, predicts that the location shown is correct (see the following subsection). Only six of the nine homologs of HSIV had an alignment to rabbit with the required coverage and E-value cutoff. The three homologs of HSIV (length 21) that do not align with 80% coverage to the rabbit sequences do, however, have perfect alignments of length 16 to the putative binding site in rabbit. The alignments cover the core binding motif, and attain an E-value of 0.001.
For both assemblies, eight of the nine CNS-2(1) homologs align to the location shown in Table 2. However, six of the CNS-2(1) homologs align to a secondary location. The secondary alignment could be eliminated positionally, as it was above IL4, whereas CNS-2(1) should be below.
Ets-1 and Gata Binding Sites Placed Using multiTF
We used Mulan to align the ENCODE rabbit genomic sequences with the nine other species shown in Table 1. We then used an option on the Mulan website to pass the multiple alignment to multiTF. The multiTF algorithm uses the alignment and the TRANSFAC matrix library, version 10.6, to identify conserved transcription factor binding sites. TRANSFAC predicts the presence of a binding site for each species individually based on its genomic sequence; it does not use the multiple alignment. The multiTF program reports locations that are in conserved regions of the Mulan alignment and that are predicted by TRANSFAC to be binding sites in all 10 species. While only eight of the 19 binding sites reported by Strempel et al 9 were also reported by multiTF, we were able to locate 17 of 19 binding sites in a conserved region reported by Mulan; see Supplementary Table S6. The two exceptions were IL13P(2) and IL13P(3).
The reason that some positions were found in a conserved block by Mulan, but were not reported by multiTF, is that TRANSFAC did not report the binding site in all 10 species. For example, the elements IL4 Promoter.1 and IL4 Promoter.2 were placed by BLAST in the ENCODE sequence at the coordinates shown in Table 2. Mulan, in fact, places these coordinates within a conserved region that extends from 830030 to 830919. The multiTF program, however, does not find a conserved Ets-1 binding site within that block.
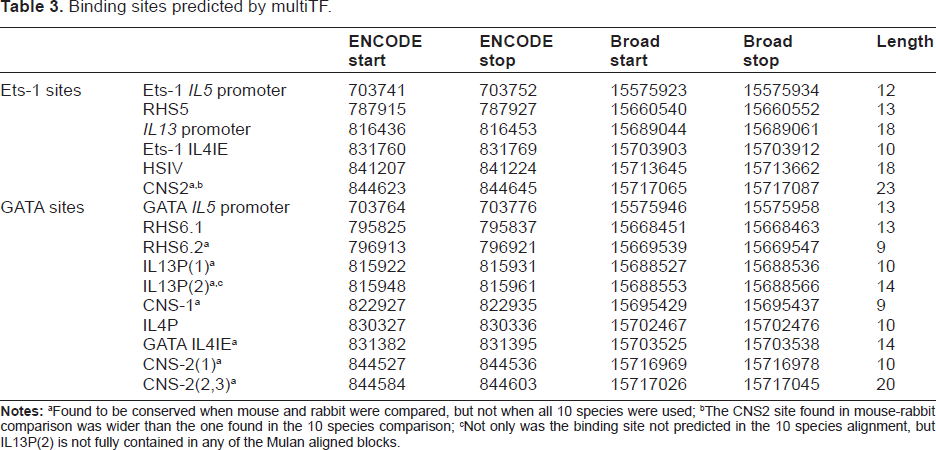
Because some of the binding sites were not predicted as conserved by multiTF in the 10-species comparison, we asked whether they were at least conserved between rabbit and mouse. We performed two more multiTF queries; one with the ENCODE genomic sequence and the mouse sequence, the other with the Broad sequence and the mouse sequence. For these queries, multiTF located 18 of the 19 binding sites from Strempel et al, 9 including the IL13P(2) site that was not in a conserved block of the 10 species alignments. These queries did not identify a conserved homolog of the IL13P(3) binding site (see next subsection). Table 3 shows the locations of the binding sites. Figure 3 shows a map of the binding sites placed relative to the IL5, RAD50, IL13, and IL4 genes on the Broad assembly.
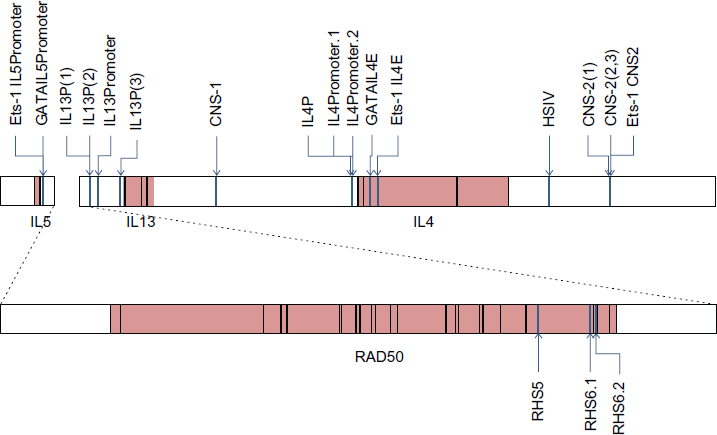
GATA and Ets-1 binding sites in Th2 region of rabbit. Diagram of the Th2 region in ENCODE assembly of rabbit, spanning rabbit sequence NT_165851.1, bases 701372 to 850370. Coordinates for genes and exons were obtained by aligning the rabbit reference mRNA sequences to the ENCODE assembly; note that two exons of IL5 were not found. The coordinates for the TFBS are as computed in this document, Table 3 IL13P(3) is included in the figure, though it is not predicted to be a binding site in rabbit. Blue lines represent binding sites, pink boxes are genes and black boxes are exons within gene. Arrows point to binding sites, so the color information is redundant. Because the RAD50 gene is large, the gene and surrounding intergenic region from bases 704372 to 850370 are drawn separately and at an approximately 3x compressed scale.
Binding sites predicted by multiTF.
Found to be conserved when mouse and rabbit were compared, but not when all 10 species were used;
The CNS2 site found in mouse-rabbit comparison was wider than the one found in the 10 species comparison;
Not only was the binding site not predicted in the 10 species alignment, but IL13P(2) is not fully contained in any of the Mulan aligned blocks.
Comparisons of Table 2 with Table 3 show that for the elements common to both tables, the results from multiTF confirm the results from direct alignment. Small differences in extent are not relevant; the extent from Table 2 should be used. Table 3 has five entries that are not found in Table 2: CNS-1, RHS6.2, IL13P(1), IL13P(2) and GATA IL4IE. CNS-1 is the only one of the five for which multiTF predicts a conserved binding site in the 10 species comparison. The Broad and ENCODE genetic sequences were identical at the positions listed in Tables 2 or 3.
I13p(3)May Not be a Gata Binding Site in rabbit
The binding site IL13P(3) seems to be lost in rabbit. In the ENCODE sequence, start of transcription for IL13 is at 817825. In mouse, IL13P(3) is 72 bases upstream, so we assume that IL13P(3), if conserved, would be located near 817825 in the ENCODE sequence. Mulan finds a conserved block that spans bases 817397 to 818100 in the 10 species alignment. The mouse and human orthologs of IL13P(3) are located in this block and are aligned with each other. The multiTF program does not predict any conserved GATA binding sites in the rabbit sequence within this block, and indeed the alignment has a gap in the rabbit sequence near the putative binding site suggested by Mulan. The gap at this location for the NZW is supported by 29 traces. The identical gap appears in the Broad sequence, supported by 11 traces, giving further evidence that IL13P(3) is missing in rabbit.
Binding Sites for Additional Transcription factors
We used multiTF with the 10-species alignment to find putative binding sites for the transcription factors listed in Table 4. The sites predicted by multiTF are shown in Table 5. Because several transcription factors were predicted to bind to more than one site, the sites were each assigned a distinct identifier, shown in the leftmost column. The block start and block stop are the beginning and end of the ENCODE rabbit sequence in the aligned block in the Mulan alignment. Figure 4 shows the location of each site within the Th2 region.
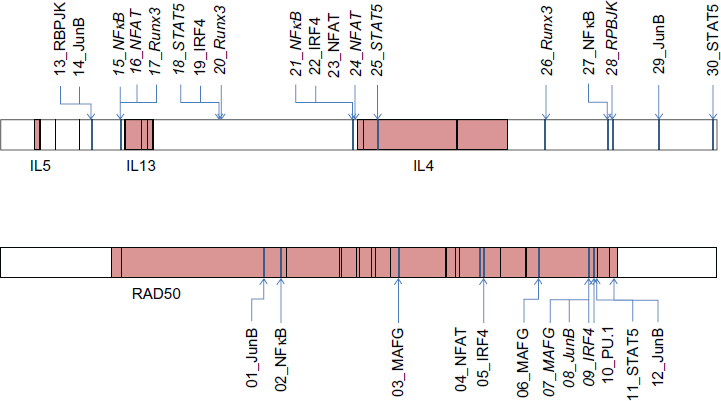
Other transcription factor binding sites in Th2 region of rabbit. Diagram of the Th2 region in ENCODE for rabbit. Coordinates, genes, and exons are the same as those used for Figure 3 The TFBS shown are the union of the sites listed in Tables 5 and 6, with sites from Table 5 shown in italicized font.
Transcription factor binding sites analyzed by comparative sequence analyses.
Transcription factor binding sites predicted in the Th2 region.
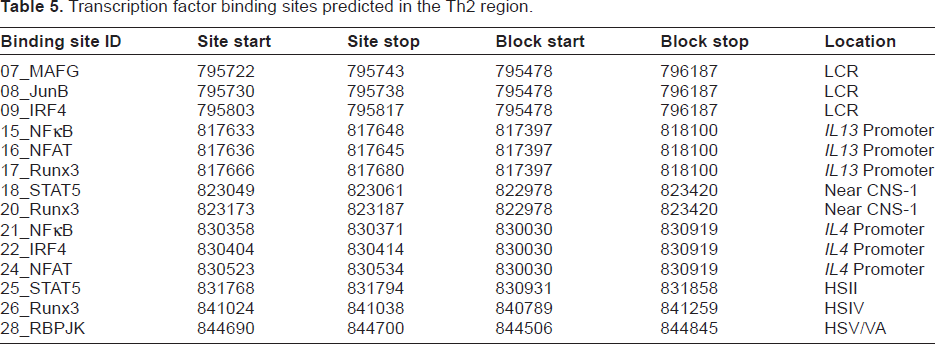
Table 5 does not contain all the sites we expected to exist in rabbit; in particular, we expected a STAT5 site in the locus control region (LCR). We sought a larger list of putative binding sites so that we could examine the Mulan alignment to determine why some of the expected sites were not found. If a site predicted by multiTF to be conserved in human, mouse and rabbit was not found in the 10-species alignment, that site was selected for further study. The sites found in the three-species alignment, but not found in the 10-species alignment, are shown in Table 6. Supplementary Tables S7–S9 show the multiple alignments for all transcription factors we studied, when such an alignment could be generated. Among the transcription factors we considered, multiTF did not predict any overlapping binding sites.
Sites found in three-species alignment for human, mouse and rabbit not found in ten-species alignment.
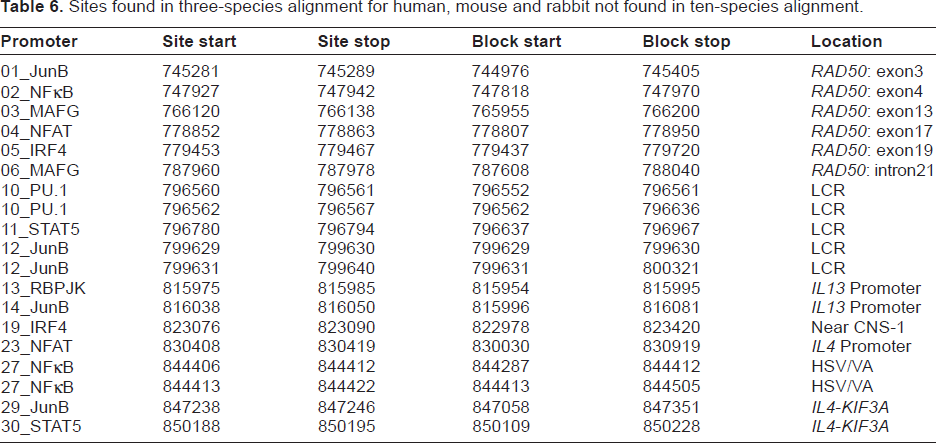
In Tables 5 and 6, sites were assigned a putative conserved noncoding region using positional reasoning described in Supplementary Data.
Discussion
The availability of two rabbit sequences for the Th2 cytokine region enabled us to do a variety of cross-species and cross-rabbit analyses. The phylogenetic study of Strempel et al 9 identified Ets-1 and GATA binding sites within major Th2 cis-regulatory elements that map to extensive (300–600 bp) regions that are highly conserved between mice and humans, but that study did not include rabbit. Because the DNA donor for the Broad 6.51x OryCun 2.0 assembly was from a partially inbred strain that had developmental defects and was more susceptible to Mycobacterial infection than outbred NZW, such as the ENCODE project's DNA donor, 2 we sought to identify differences in the exons and transcription factor binding sites that might be associated with the phenotypic differences.
As summarized in Supplementary Table S3, we found a substitution in IL13 and a frame shift in IL4 that might be relevant to the phenotypic differences. Other discrepancies in the assemblies included missing exons (IL5 in ENCODE) and extra exons (KIF3A in OryCun 2.0). That the only available full rabbit genome assembly is from an extinct strain with an abnormal phenotype poses problems for future rabbit genomic studies. The OryCun 2.0 assembly has hundreds of regions of contiguous assembled sequence that are not placed on any chromosome, many stretches with ambiguity characters (Ns) and poor coverage of some regions such as the potential frameshift in IL4.
Comparative Analyses of Binding Sites and Promoter regions
We could place 18/19 Ets-1 and GATA binding sites described in Strempel et al 9 on the Broad OryCun 2.0 assembly and the ENCODE rabbit region ENm002. The sequence that was not placed was identified by Strempel et al 9 as a GATA binding site, but was not predicted to be a GATA binding site in rabbit. The rabbit sequence does align to the orthologous binding sites, but there is a single nucleotide deletion in rabbit, causing the rabbit sequence not to be predicted as a GATA binding site. Twelve of 19 sequences could be directly placed using BLAST, and so are highly likely to be identified correctly. Six others could be placed by multiple alignment, plus a prediction of transcription factor binding sites by multiTF. There were binding sites that could only be placed by BLAST and were not predicted by multiTF.
We conducted analyses of additional transcription factor binding sites. Among the sites that were not conserved across all species, the 11_STAT5 site particularly caught our attention because this TFBS is located in the locus control region. The Papio anubus sequence was not predicted to have a STAT5 binding site at the homologous location, and there were 19 traces in the trace archive that support the Papio anubus sequence. Callithrix jacchus has a gap in the alignment where the STAT5 binding site would be. More generally, the “no” entries in Supplementary Table S9 define a set of (species, transcription factor, site) combinations that merit further investigation. If these sites are not present in all mammals, then this would have implications for evolution of T cell regulation.
Roles for rad50 and KIF3A
This study includes analysis of RAD50 and KIF3A, although they do not encode Th2 cytokines. Why are these genes conserved in syntenic relationships to the cytokine genes, including avian species thought to have diverged from the mammalian lineage 300 million years ago? Although RAD50 is widely expressed, it appears to serve a secondary function in its location by harboring locus control sequences in its 3’ untranslated region.17,22,23 Locating the LCR within RAD50 but near the IL4 and IL13 cytokine genes may be advantageous because RAD50 is accessible and transcribed as a housekeeping gene and at the same time, the LCR contributes to the regulation of the adjacent IL4 and IL13. Similarly KIF3A may be preserved in the syntenic relationship to serve secondary functional roles. There is complex epigenetic control of the polarization steps toward characteristics of activated Th2 cells.24–27 Chromatin remodeling brings together distant sites within the locus. 28 Th2 cell activation upregulates production of SATB129,30 which then binds to CNS1, CNS2 and 9 other sites extending from IL5 past KIF3A. CTCF also binds between IL5 and the neighboring IRF1 and within the KIF3A gene, helping to segregate the Th2 domain from surrounding regions. 31
Tuberculosis and the Th2 Cytokine region
At the start of this project, we hypothesized that it was possible that variants in the region with IL4, IL13, and other nearby genes of immunological interest (IL5, RAD50) could have contributed to some immune system deficits in the partially inbred Thorbecke rabbit that led to this strain's decreased resistance to tuberculosis. 2 Recently, in a family-based association study of human tuberculosis, potential risk haplotypes contributing to tuberculosis susceptibility were suggested to reside on a region of human chromosome 5 encompassing Th2 cytokines within a three-marker haplotype of SNPs in SLC22A4, SLC22A5 and KIF3A. 32 This haplotype may influence cytokine expression levels and influence the magnitude of T-cell responses to Mycobacterium tuberculosis.
The sequence differences we found between the Broad and ENCODE assemblies appear to be largely due to N's in the Broad assembly or sequencing errors, not true differences in the DNA sequences of the two donor rabbits. A splice variant equivalent to human IL4δ2 33 was reported in rabbits in 2000, 34 in several primates 35 and in mice. 36 There is a possible frameshift mutation in exon 2 of the IL4 gene in the rabbit used for the Broad assembly. If correct, this could force production of the alternatively spliced IL4δ2 variant product that lacks exon 2, at least from one allele. A pathological role of IL4 and other type 2 cytokines during responses to pulmonary infections with Mycobacterium tuberculosis has been suggested. 5 Although patients with increased expression of IL4 mRNA had more extensive disease, they were also observed to exhibit greater expression of IL4δ2. 3 Accurate measurements of message levels are complicated by relative instability of IL4δ2 message. 4 In addition, determinations of mRNA expression levels in cells obtained from sites of infection may be more relevant than measurements of levels produced by cells from peripheral blood.4,5 The rabbit is an excellent model for human pulmonary tuberculosis because lung pathology in both man and rabbit includes pulmonary granulomas with caseous necrosis. 2 Increased IL4 production in tuberculosis was associated with development of pulmonary cavities.37,38 Recently Luzina et al reported differences in pulmonary cytokines and cellular infiltrates elicited when human or murine full-length IL4 or IL4δ2 was virally expressed in mouse lungs.39,40 Their studies demonstrate functional roles for IL-4δ2 independent and distinct from IL4. Even if the possible frameshift were a sequencing error, further studies of Mycobacterium tuberculosis models in rabbits should evaluate IL4 levels and screen for expression levels of both the long and IL4δ2 forms of IL4.
Our sequence analysis of the Th2 region in rabbit and other mammals suggests areas for further investigation in at least four directions. First, the transcription factor binding sites in the Th2 region appear to be variably conserved in mammalian evolution. The immunological function of binding sites present in some mammals and absent in others should be tested. Second, there are likely sequence differences between rabbits in the exons of Th2 region genes. Third, laboratories currently using the rabbit model do observe different responses to experimental infection with M. tuberculosis.41,42 Potential differences in binding sites and in the coding regions of IL4 and IL13 reported here may be confirmed and extended in future studies using rabbits that develop differential disease presentation when infected with the same species and strain of Mycobacterium. Finally, deficiencies in the assembly and annotation of the current OryCun2.0 rabbit genome sequence emphasize the need for further sequencing of rabbits from other strains of this species and improved assembly of the many complex regions of interest to immunologists.
Methods
Ets-1 and Gata Binding sites
The supplemental data found in Strempel et al 9 provided the genomic sequences of 19 evolutionarily conserved sites in the nine species listed in Table 1. One hundred seventy, rather than 171, sequences are listed because no sequence for the RHS 6.2 site was available for Callithrix jacchus. Ten of 19 sites have sequences length 14, eight have length 21, and one has length 25. Eight of the listed sites are Ets-1 binding sites, and 11 are GATA binding sites.
Alignments Using BLAST
We used NCBI BLAST 20 to align the sequences of the 170 binding sites to the Broad OryCun 2.0 and ENCODE rabbit sequences cited in Table 1. We used version 2.2.23 of BLAST with word size 4, match reward 2, mismatch penalty -3 and no filtering (options: -r 2 -q -3 -W 4 –FF). In our usage, the purpose of using –FF was to show that even with filtering off, multiple placement was not a problem. For some results, we filtered the BLAST output further. We excluded alignments with less than 80% coverage of the query sequence. Coverage is defined as the extent of the alignment in the query, divided by the full length of the query. We also applied a filter to the BLAST results that excluded alignments with E-value. 0.1. We did not use the –E option to BLAST because this option affects some of the internal heuristics.
Multi-Species alignment
We used the Mulan 21 alignment algorithm (http://mulan.dcode.org/), to align the genomic sequences shown in Table 1. We generated four distinct alignments using Mulan: one that aligned the ENCODE sequence with the sequences from the other nine species; one that aligned the ENCODE sequence with the syntenic region from mouse; one that aligned the Broad OryCun 2.0 sequence to the syntenic sequence from mouse; and one that aligned human, mouse, and the ENCODE sequence for rabbit.
Prediction of Binding Sites Using multiTF
We used multiple alignments produced by Mulan as input to multiTF (http://multitf.dcode.org/), a program that uses the TRANSFAC 10.6 library to identify conserved transcription factor binding sites.
The binding factors in the TRANSFAC 10.6 library with identifiers beginning with “V$CETS” or “V$ETS”, except for “V$ETS2_B”, were considered to identify Ets-1 binding sites. The binding factors with identifiers starting with “V$GATA” were considered to identify GATA binding sites.
We sought binding sites for the additional transcription factors listed in column one of Table 4. The matrices used by multiTF to recognize the transcription factor binding sites are shown in the second column. Putative binding sites were found in the Th2 region for all the matrices listed in Table 4, except for the two matrices marked with an asterisk. Both of these would recognize binding sites for STAT5. The V$ETS_Q6 matrix, which recognizes PU.1, also recognizes the transcription factor Ets-1, so we filtered known Ets-1 binding sites from the list of predicted PU.1 binding sites.
Strempel et al 9 warn of incompleteness of the Callithrix jacchus sequence near the Th2 locus control region. We found no specific case in which the Callithrix jacchus genome alone prevented recognition of a binding site for one of the transcription factors listed in Table 4. Therefore, we did not handle Callithrix jacchus in any special way.
Acknowledgements
This research was supported by the Intramural Research Program of the National Institutes of Health, NLM and NIAID. Thanks to David Margulies for advice on using PyMol. Thanks to Jinfang Zhu for helpful suggestions on the design of the study and on the text of the manuscript. We also appreciate additional comments on the manuscript from Alan Sher, Michael Mage, and Laura Via.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Supplemental Material
Alignment of reference sequences to the rabbit genomic regions
We used the Splign 1 program to align the reference mRNA sequences shown in Table S1 to the full length of the Broad and ENCODE genomic sequences summarized in Table 1. For both assemblies, the placement derived from this alignment is shown in Table S2. The alignment almost entirely confirmed the exon placement recorded in the Entrez Nucleotide record of the Broad sequence, except for exon 6 of RAD50, which is explained below. This confirmation is not surprising, as the placement recorded in the database was derived from a similar alignment. IL5 could not be placed by Splign in the ENCODE sequence, but exons 1 and 2 could be placed by BLAST, and the locations of these exons are shown in the Table S2.
To provide cross-species support for the placement of coding exons, we used TBLASTN 2 with low-complexity filtering disabled (option –FF) to align the human protein sequences listed in Table S1 to both rabbit assemblies. The alignments mostly confirm the placement of the exons found by rabbit mRNA alignment, with occasional differences in length as expected for cross-species comparison (data not shown). The alignments did not confirm the placement of exon 6 of RAD50 on the Broad assembly, and did confirm the absence of IL5 exons 3 and 4 in the ENCODE genomic sequences. Furthermore, there are no exons of human KIF3A that correspond to exons 10 and 11 of rabbit reference mRNA.