
Abstract
Highly active antiretroviral (ARV) therapy (HAART) for chronic suppression of HIV replication has revolutionized the treatment of HIV/AIDS. HAART is no panacea; treatments must be maintained for life. Although great progress has been made in ARV therapy, HIV continues to replicate in anatomical and intracellular sites where ARV drugs have restricted access. Nanotechnology has been considered a platform to circumvent some of the challenges in HIV/AIDS treatment. Dispersion polymerization was used to fabricate two types (PMM and ECA) of polymeric nanoparticles, and each was successfully loaded with four ARV drugs (zidovudine, lamivudine, nevirapine, and raltegravir), followed by physicochemical characterization: scanning electron microscope, particle size, zeta potential, drug loading, and in vitro availability. These nanoparticles efficiently inhibited HIV-1 infection in CEM T cells and peripheral blood mononuclear cells; they hold promise for the treatment of HIV/AIDS. The ARV-loaded nanoparticles with polyethylene glycol on the corona may facilitate tethering ligands for targeting specific receptors expressed on the cells of HIV reservoirs.
Introduction
Highly active antiretroviral (ARV) therapy (HAART) for chronic suppression of HIV replication has revolutionized the treatment of HIV/AIDS: many patients are now in their second decade of treatment, with levels of plasma HIV RNA below the limits of detection of clinical assays. 1 However, HAART is no panacea. Current treatments must be maintained for life. Although great progress has been made in ARV therapy, HIV continues to replicate in anatomical and intracellular sites where ARV drugs have restricted access. 2 Thus, there is a great need for continued efforts in seeking innovative approaches for the treatment of HIV/AIDS. Cellular and anatomical reservoirs of HIV-1 represent major impediments to eradication. 3 Cellular reservoirs include latently infected CD4+ T cells, macrophages, and follicular dendritic cells. The main anatomical reservoir sites for HIV include the lymphoid organs (particularly the gut-associated lymphoid tissue [GALT]) and the brain. 3 They are the sites of persistent HIV replication. HIV is active in the GALT throughout the period of clinical latency, although minimal viral burden is observed in the blood. 4 6
Viral replication occurs predominantly in the peripheral lymphoid organs, and the GALT is an important site of active viral replication, possibly reflecting high levels of T-cell activation.3,7–10 The maintenance of viral reservoirs revolves around the GALT of HIV-infected individuals despite long-term viral suppression.2,11 The belief is that the ongoing low level of replication in the GALT may replenish the cellular reservoir. 12 Given the restricted access of HAART to anatomical reservoirs, nanotechnology has been considered a platform that can be brought to bear to circumvent some of the challenges in HIV/AIDS treatment. Nanomedicine platforms that deliver ARV agents to HIV reservoirs (drug polymer conjugates, dendrimers, micelles, liposomes, solid nanoparticles, nanosuspensions, polymeric nanoparticles, and cell-mediated nanoparticles) have been discussed recently. 2 Polymeric nanoparticles have higher drug-loading capacity and the potential to target HIV reservoirs by manipulation of their surface characteristics. 13 15 Polymeric nanoparticles are preferentially taken up by the M cells of Peyer's patches of the GALT. 16 Although M cells possess the capability to transcytose synthetic nanoparticles, the absorption occurs at very low levels. Thus, there is scope for the application of the concept of site-specific targeting of ARV drug-loaded nanoparticles to M cells to enhance the efficacy of orally administered ARV drugs.
We believe that grafting an M-cell-targeting ligand to the nanoparticle surface could improve M-cell oral delivery strategies by influencing the endocytic mechanisms involved in their uptake by M cells.
17
22
Given the report that the ongoing low level of replication in the GALT may replenish the latent reservoir,6,12 this series of projects will result in targeted ARV drugs-loaded nanoparticle delivery system to facilitate uptake and transcellular transport of the ARV drugs to minimize GALT reservoir responsible for the persistent ongoing replication of HIV-1 (ie, adapting drug delivery system properties to cell requirements), as shown in Scheme 1. We have used dispersion polymerization (in situ polymerization) technique previously for the fabrication of nanoparticles for the delivery of bioactive agents.
23
26
In situ polymerization of monomers, including cross-linkers in the fabrication of polymeric nanoparticles, allows one-pot synthesis of nanoparticles with many advantages.
26
All the advantages of in situ polymerization derive from the possibility of simultaneous encapsulation of bioactive agents during polymerization and, by copolymerization, the possibility of adding surface functionalities in one-batch process without further modifications compared to nanoparticle fabrication by dispersion of preformed polymers.
23
27
Our in situ polymerization technique involves the use of a macromonomer or a monomer that forms the core of the nanoparticles, a cross-linker that cross-links the core, a redox initiator system, and a hydrophilic macromonomer (polyethylene glycol [PEG]), which confers the stealth property to the nanoparticles and which also serves as a stabilizer. We have developed polymerizable macromonomers bearing a site-specific ligand (GRGDS peptide) for the receptor expressed uniquely on the apical surface of M cells (cell surface β1 integrin) using low molecular weight (MW) polydisperse polyethylene glycol methacrylates
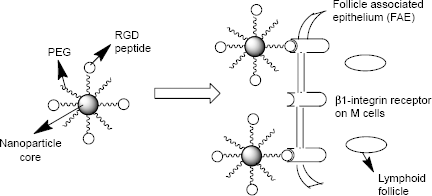
Surface-modified nanoparticles capable of crossing the intestinal epithelium via β1-integrin receptors on M cells.
Here, we fabricated polymeric nanoparticles that were loaded with four different ARV drugs: zidovudine (nucleoside reverse transcriptase inhibitor), lamivudine (nucleoside reverse transcriptase inhibitor), nevirapine (nonnucleoside reverse transcriptase inhibitor), and raltegravir (HIV integrase strand transfer inhibitor). We tested encapsulation efficiency and release of ARV drugs and the effect of ARV drugs-loaded nanoparticles on T cells and macrophages during one-round HIV-1 infection.
Materials and Methods
Materials
Zidovudine, lamivudine, and nevirapine (Sigma-Aldrich), raltegravir (APAC Pharmaceutical, LLC), hydroxylamine hydrochloride (99%; Sigma-Aldrich), pyridine (99+%; Sigma-Aldrich), diethyl ether (99.8%; Sigma-Aldrich), ethyl alcohol (denatured; Sigma-Aldrich), methyl alcohol (99.93%; Sigma-Aldrich), acetonitrile (99.93+%; Sigma-Aldrich), and hydrochloric acid (37%; Fisher Scientific) were used as received. Methacryloyl chloride (97%; Sigma-Aldrich) and methyl methacrylate (99%; Sigma-Aldrich) were vacuum distilled prior to use. Poly(ethylene glycol)
Cells and Media
CD4+ T cells (CEM) were obtained from the NIH AIDS Research and Reference Reagent Program. Peripheral blood mononuclear cells (PBMCs) were purchased from Astra Biologics. Cells were cultured at 37°C in a 5% CO2 atmosphere in RPMI 1640 medium (Invitrogen) containing 10% fetal bovine serum and 1% antibiotic solution (penicillin and streptomycin; Invitrogen). PBMCs were stimulated with 2.5 mg/mL phytohemagglutinin for 24 hours and then activated for another 24 hours with 10 units/mL interleukin 2 before the infection with vesicular stomatitis virus g-glycoprotein (VSVg)-pseudotyped HIV-1 virus expressing luciferase (HIV-1 Luc).
Methods
Synthesis of Hydrolyzable Cross-Linker (N,O-Dimethylacry-Loylhydroxylamine)
The cross-linker,
Synthesis and Characterization of Methacrylate-Terminated Poly-ϵ-Caprolactone Macromonomer
The synthesis was done as reported previously. 26 ϵ-Caprolactone (13.14 g), HEMA (4.0 g), and SnOct (three drops) were placed in a 50-mL round-bottom flask equipped with a magnetic stir bar. The reaction was kept under vacuum for 10 minutes, and polymerization was carried out in an inert atmosphere by flushing the flask with nitrogen gas for 24 hours on a silicone oil bath kept at 128°C. The resulting macromonomer was dissolved in dichloromethane and filtered. The macromonomer was precipitated out of this solution using an excess amount of hexane and dry ice. Then, the material was filtered and dried in a vacuum oven at room temperature under reduced pressure over phosphorus pentoxide for 24 hours. The FT-IR and 1 H NMR spectra of the synthesized macromonomer were consistent with the expected structure.26,31,32 The number average MW (Mn) was determined by 1 H NMR and gel permeation chromatography (GPC), while the weight average MW and polydispersity index of the macromonomer were determined by GPC using a Waters 2690 GPC system equipped with a Waters 2410 differential refractive index detector. 24 26 Polystyrene standards were used for calibration and tetrahydrofuran was used as the mobile phase.
Synthesis of Nanoparticles
Methyl Methacrylate-Based Nanoparticles (Polymethyl Methacrylate [PMM], Scheme 2)
8 mmol of methyl methacrylate, 0.1 mmol of PEG–methyl methacrylate (PEG-MMA) macromonomer, and 0.162 mmol of the cross-linker (
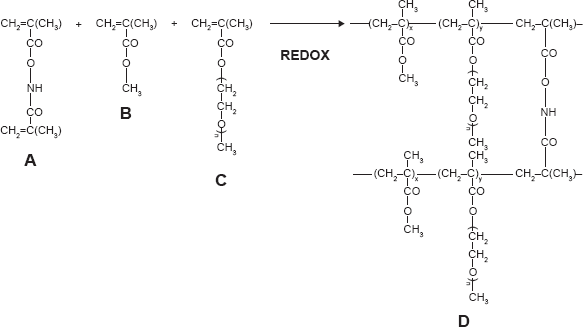
Synthesis and structure of stealth hydrolyzable crosslinked PEG-PMMA (PMM) nanoparticles (A =
ϵ-Caprolactone-Based Nanoparticles (ECA; Scheme 3)
0.24 mmol of poly-caprolactone-HEMA macromonomer and 0.5 mmol of PEG-MMA, with 0.0209 mmol of cross-linker, were dispersed in acetone:water (10 mL). 0.3325 mmol of BP and 0.3325 mmol of NDPEA (as co-initiators) were injected through the rubber closure under continuous flushing with nitrogen gas and with continuous stirring at 400 rpm. Ten milligrams of each of the drugs (zidovudine, lamivudine, nevirapine, and raltegravir) were added. The overall polymerization time was 24 hours. The nanoparticles were recovered by centrifugation and then lyophilized.
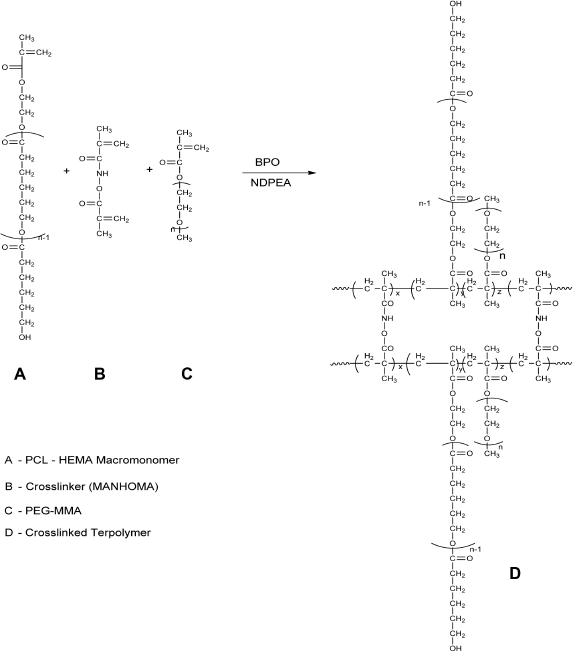
Synthesis and structure of stealth crosslinked poly-ϵ-Caprolactone-HEMA Nanoparticle (ECA).
Characterization of the Nanoparticles
Surface Morphology
The morphology of the nanoparticles was investigated using scanning electron microscope (SEM; JSM-5300 SEM; JEOL). The freeze-dried particles were fixed on a double-sided sticky carbon tape. Then, they were sputter coated with gold in a Denton Vacuum Inc. Desk II instrument (45 mA) and examined in the JSM-5300 SEM instrument at 15 kV.
Particle Size Analysis and Zeta Potential
Particle size and size distribution and zeta potential measurements of the nanoparticles were performed by photon correlation spectroscopy using Zetasizer® 3000 (Malvern Instruments). The measurement was performed at 25°C. The dried nanoparticles were suspended in filtered distilled water (3 mL) by vortexing for one minute on a touch mixer (Model 231; Fisher Scientific) and then sonicated for three minutes using a Sonics Vibracell™ Amplitude sonicator (Sonics &Materials, Inc.) before measurement.
Drug Loading and in vitro Drug Availability
Preparation of Calibration Curves for Zidovudine, Lamivudine, Nevirapine, and Raltegravir
Hewlett Packard 1100 Series, a high-performance liquid chromatography (HPLC) system, was used. Separation was done at 35°C on Eclipse Plus C18 column (4.6 mm X 150 mm) from Agilent Technologies. The mobile phase comprised solution A (0.01 M KH2PO4) and B (acetonitrile). The injection volume was 20 μL. The analysis was done at gradient mode with a flow rate of 1 mL/minute; the ratio A/B changed from 90/10 to 20/80 gradually in five minutes and readily reequilibrated to 90/10. Total analysis time was eight minutes. The stock solution of raltegravir, nevirapine, zidovudine, and lamivudine (1 mg/mL) was prepared by dissolving 5 mg of each drug in 5 mL of methanol. Then, the stock solution was diluted with phosphate-buffered saline (PBS) for the preparation of the working solutions as follows: 0.025, 0.05, 0.1, 0.25, 0.5, 1.0, 2.5, 5.0, 10.0, 25.0, 50.0, and 100.0 μg/mL. Ultraviolet–visible detection was performed using an HP photodiode array detector. Blank samples showed no interfering substances eluting at the retention time of raltegravir (retention time, 5.7 minutes; detected at 310 nm), nevirapine (retention time, 4.6 minutes; detected at 285 nm), lamivudine (retention time, 2.7 minutes; detected at 267 nm), and zidovudine (retention time, 3.7 minutes; detected at 267 nm). Furthermore, linear calibration curves (peak areas versus drug concentrations) were established for raltegravir, nevirapine, zidovudine, and lamivudine over 0.05–100.0 μg/mL concentration range (
In vitro Availability Studies
Drugs (raltegravir, nevirapine, zidovudine, and lamivudine)-loaded nanoparticles (known weight) were suspended in PBS (pH 7.4; IS 0.16), placed in a dialysis bag (membrane tubing with MW cut-off of 12,000–14,000) in centrifuge tubes containing known volume of PBS (pH 7.4; IS 0.16), and rotated at 360° using Labquake Shaker maintained at 37°C as reported previously. 23 26 At specific time intervals, an aliquot of the release medium was taken and replaced with a fresh medium (maintained at 37°C) each time to keep the volume constant. Then, the aliquot release medium was filtered through a 0.45-μm syringe filter, and the amount of drugs (raltegravir, nevirapine, zidovudine, and lamivudine) released into the medium was quantified using a validated HPLC method (quantitation was carried out according to the standard curves prepared for the drugs).
Determination of Drugs-Loading Capacity of the Nanoparticles
The determination was done as reported previously
26
The HPLC analysis was the same as used for the in vitro availability studies of the drugs-loaded nanoparticles described earlier. The nanoparticles (ANP) were incubated in the release medium for more than 35 days till all the loaded drugs were released (each sample in triplicate). Then, quantitation of each drug (raltegravir, nevirapine, zidovudine, and lamivudine) was carried out using the standard curves. Encapsulation property of the nanoparticles was expressed as the loading capacity (amount of drug encapsulated expressed as a percentage of the nanoparticles' weight), using the following equation:
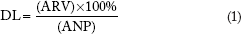
VSVg-Pseudotyped HIV-1 Luc
The 293T cells were cotransfected with HIV-1 proviral vector pNL4-3.Luc.R-E- and VSVg-expressing vector (both obtained from the NIH AIDS Research and Reference Reagent Program). At 48–72 hours post transfection, the media containing HIV-1 virions were collected, filter-sterilized, and kept frozen. HIV-1 Luc was added to CEM T cells or PBMCs. After 48 hours, the cells were washed in PBS and lysed with Steady Lite Luciferase buffer (PerkinElmer). Light emission was analyzed in Luminoskan (PerkinElmer). To adjust for the cell number, the samples were counted with trypan blue as described in the following section.
Cell Viability Assays
Cells were cultured in 96-well plates at 37°C and incubated with nanoparticles at the indicated concentrations for 24 hours. Cell viability was determined using trypan blue-based assay. The cells were supplemented with 0.2% trypan blue, transferred to a plastic disposable counting chamber, and counted on Cellometer Automatic Cell Counter (Nexcelom Bioscience).
Quantitative PCR for Viral DNA Analysis
PBMCs were infected with HIV-1 Luc and treated with nanoparticles for six hours. Total DNA was extracted from 4 X 106 cells using lysis buffer (10 mM Tris-HCl pH 8, 10 mM EDTA, 5 mM NaCl, and 200 μg/mL proteinase K). The cells were lysed for 20–30 minutes at room temperature, and proteinase K was inactivated by heating to 95°C for five minutes. For the real-time PCR analysis, 100 ng DNA was amplified in Roche LightCycler 480 II (Roche Diagnostics) and SYBR Green Master mix (Roche Diagnostics). PCR was carried out with initial preincubation for five minutes at 45°C and then for three minutes at 95°C followed by 45 cycles of denaturation at 95°C for 15 seconds, annealing and extension at 60°C for 45 seconds, and final extension at 72°C for 10 seconds. Quantification of an early LTR was carried out using β-globin DNA as a normalization standard. Primer sequences for early LTR were forward-GGCTAACTAGGGAACCCACTG and reverse-CTGCTAGAGATTTTCCACACTGAC and for β-globin were forward-CAACCTCAAACAGACAC-CATGG and reverse-TCCACGTTCACCTTGCCC (for primer information).
33
Mean Cp values for early LTR and β-globin were determined, and AACT method was used to calculate the relative expression levels. Unpaired
Results and Discussion
The data obtained from the synthesis of the cross-linker
Two types of nanoparticles were synthesized using dispersion polymerization technique: stealth methyl methacrylate-based (PMM; Scheme 2) and stealth ϵ-caprolactone-based (ECA; Scheme 3) nanoparticles. Methyl methacrylate monomer (Scheme 2) or poly-ϵ-caprolactone macromonomer (Scheme 3), PEG-MA (
Morphological Evaluation of the Nanoparticles
Nanoparticle fabrication by dispersion polymerization was confirmed by SEM for the two types of nanoparticles (stealth methyl methacrylate based [Scheme 2] and stealth ϵ-caprolactone based [Scheme 3]). Scanning electron micrographs of ARV drugs-loaded hydrolyzable cross-linked PEG–poly-ϵ-caprolactone nanoparticles (Fig. 1A and 1B) and ARV drugs-loaded hydrolyzable cross-linked PEG–polymethyl methacrylate nanoparticles (Fig. 1C) show the formation of smooth spherical nanoparticles.

Scanning electron micrographs of ARV drugs-loaded hydrolyzable crosslinked PEG-poly caprolactone (ECA) nanoparticles (
The average particle size for cross-linked PEG–polycaprolactone nanoparticles (Fig. 2A) is 321 ± 36 nm with 0.344 ± 0.023 PDInanoparticle, while the average particle size for cross-linked PEG-PMM nanoparticles (Fig. 2B) is 137.9 ± 1.3 nm with 0.280 ± 0.0.021 PDInanoparticle. Thus, the nanoparticles obtained with poly-ϵ-caprolactone as the hydrophobic core are bigger than the nanoparticles obtained with PMM as the hydrophobic core. The particle size distribution is given by the PDInanoparticle (a PDInanoparticle value of ≥0.1 indicates a homogenous monodisperse formulation, while a PDInanoparticle of >0.3 indicates polydispersity with large variations in particle size). 24 The PDInanoparticle values obtained in this work indicate that PEG-PMM nanoparticles are more homogenous than PEG–poly-ϵ-caprolactone nanoparticles. The zeta potential on the surface of PEG–polycaprolactone-based nanoparticles is more negative (indicating greater tendency for colloidal stability: −31.4 ± −3.6 mV) than the zeta potential on the surface of PEG-PMM-based nanoparticles (-19.2 ± −2.8 mV). It has been reported that particle size plays an important role in determining drug release behavior of drug-loaded nanoparticles and their fate after in vivo administration. 34 Furthermore, the zeta potential results obtained in this work show predominantly negative values. Following administration, nanoparticles with a positive zeta potential pose a threat of causing transient embolism and rapid clearance compared to negatively charged particles. Thus, the pegylated poly-ϵ-caprolactone-based nanoparticles and pegylated PMM-based nanoparticles fabricated by dispersion polymerization are very promising: capable of carrying the drug load to its site of action. 35
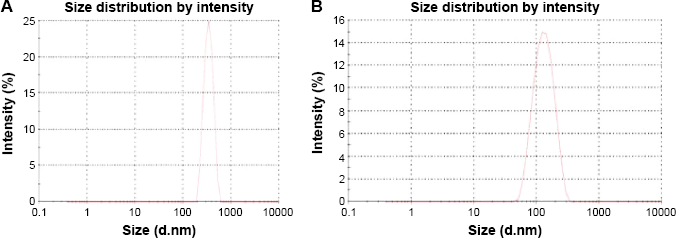
Typical particle size distribution for PEG-polycarpolactone (
Drug-Loading Capacity and in vitro Availability of ARV Drugs from Nanoparticles
Drug-loading capacity was determined by a validated HPLC method as described previously and found to be 6.44 ± 0.13% w/w (raltegravir), 2.13 ± 0.25% w/w (lamivudine), 0.88 ± 0.16% w/w (zidovudine), and 4.15 ± 0.39% w/w (nevirapine) for PEG-PMM-based nanoparticles. For PEG–polycaprolactone-based nanoparticles, the results are as follows: 6.79 ± 0.8% w/w (raltegravir), 1.02 ± 0.12% w/w (lamivudine), 2.11 ± 0.38% w/w (zidovudine), and 5.13 ± 0.58% w/w (nevirapine). While the drug-loading capacity values for each of the drug are better than the values obtained for paclitaxel loaded in PEG–poly(L-lactide) 25 and docetaxel loaded in PEG–poly(ϵ-caprolactone) nanoparticles 26 (prepared by dispersion polymerization), which are 0.25% and 0.8%, respectively, effort will be made to improve the loading capacity.
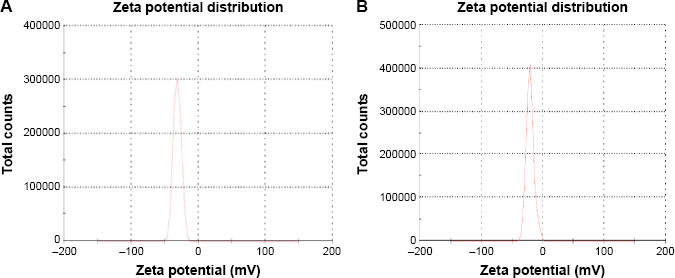
Typical particle zeta potential distribution for PEG-polycarpolactone (
Figures 4 and 5 show the in vitro release isotherms of the ARV drugs from PEG-PMM-based and PEG–polycaprolactone-based nanoparticles, respectively. The release is biphasic with an initial rapid release followed by a slower rate of release. Drug-loading capacity and in vitro availability of the drugs from the nanoparticles appear to be independent of the type of nanoparticles (PEG-PMM-based or PEG–polycaprolactone-based nanoparticles). It appears that the physicochemical properties of the drugs control drug loading and drug release. Raltegravir used in this work (MW: 443.42 g/mol) exhibits poor aqueous solubility (ie, ≥1 mg/mL); it is very slightly insoluble according to the classification of Amidon and his group, 36 whereas the potassium salt of raltegravir is significantly more soluble in water. 37 As shown in Figures 4 and 5, raltegravir-loaded nanoparticles, with the highest drug-loading capacity, released the highest amount of drug, followed by nevirapine (MW: 266.31 g/mol), lamivudine (MW: 229.26 g/mol), and zidovudine (MW: 267.4 g/mol). Based on the data from NIH AIDS Research and Reagent Program (Reagent Information, September 2011), 38 the rank order of their aqueous solubilities is as follows: nevirapine (0.1 mg/mL: very slightly insoluble in water) > zidovudine (20 mg/mL: sparingly soluble) > lamivudine (70 mg/mL: soluble). Thus, the more hydrophobic ARV drugs have a greater affinity for the hydrophobic cores of the nanoparticles; they exhibit high drug-loading capacity and high drug release.
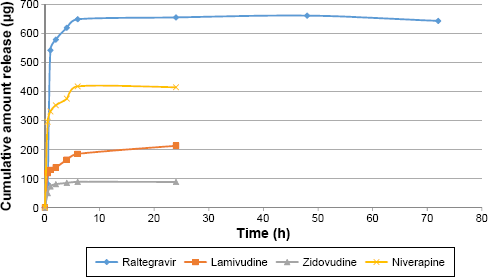
Cumulative amount released versus time for ARV drugs-loaded PEG-polymethylmethacrylate based nanoparticles (Raltegravir: ϵ = 8.15–31.28 μg; Niverapine: ϵ = 7.5–36.84 μg; Lamivudine: ϵ = 1.20–3.59 μg; and Zidovudine: ϵ = 2.06–16.74 μg).
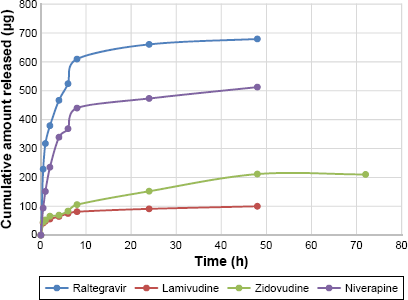
Cumulative amount released versus time for ARV drugs-loaded PEG-polycaprolactone based nanoparticles (Raltegravir: ϵ = 6.31–30.2 μg); Niverapine: ϵ = 6.7–57 μg; Lamivudine: ϵ = 0.14–4.71 μg; and Zidovudine: ϵ = 0.56–37.46 μg).
HIV-1 Inhibition Experiments
ARV drugs-loaded stealth PMM-based nanoparticles (containing raltegravir, lamivudine, zidovudine, and nevirapine) inhibit one-round HIV-1 infection in cultured T cells and primary PBMCs (Fig. 6). The ARV-loaded PMM-based nanoparticles inhibited HIV-1 infection in CEM T cells by more than 90% with low micromolar EC50 (Fig. 6A). We next tested the effect of the PMM nanoparticles on one-round HIV-1 infection of PBMCs. The ARV-loaded but not blank nanoparticles markedly inhibited HIV-1 replication with EC50 = 14 μM (Fig. 6B). To confirm that the mechanism of HIV-1 inhibition by the ARV-loaded PMM nanoparticles was due to the inhibition of HIV-1 reverse transcription (RT), we analyzed early RT by quantifying HIV-1 DNA. 33 The established HIV-1 inhibitor, zidovudine, inhibited RT (Fig. 6C). The ARV-loaded PMM nanoparticles (10 μM) inhibited RT, while blank nanoparticles had little effect (Fig. 6C), suggesting that HIV-1 inhibition was due to the reduction of RT. To access the toxicity of PMM-based nanoparticles, PBMCs were treated with different concentrations of PMM-based nanoparticles and a trypan blue exclusion assay was performed to assess cell's viability. The assay showed that the ARV-loaded PMM-based nanoparticles were toxic at higher concentration (CC50 = 42 μM; Fig. 6D).
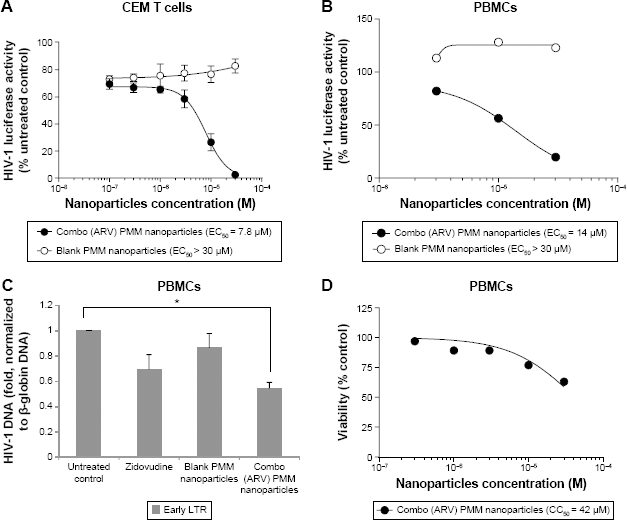
Inhibition of HIV-1 one round infection by PMM nanoparticles. (
HIV-1 Inhibition by ARV Drugs-Loaded Stealth Polycaprolactone (ECA) Nanoparticles
We tested ARV-loaded ECA nanoparticles for HIV-1 inhibition. In CEM T cells infected with HIV-1 Luc, the combo (ARV) ECA-based nanoparticles achieved only ~25% inhibition with EC50 = 0.07 μM (Fig. 7A). This effect was not due to toxicity as nanoparticles were not toxic at this concentration (CC50 = 0.8 μM; Fig. 7B). Data were not available for blank nanoparticles as we could not disperse them well enough in culture media, possibly due to a low level of PEG on nanoparticle surface. The ARV-loaded ECA nanoparticles (10 μM) inhibited HIV-1 RT in the infected PBMCs similar to zidovudine (Fig. 7C). This was not due to toxicity as the ARV-loaded PMM-based nanoparticles were toxic at higher concentrations (CC50 = 25 μM; Fig. 7D).
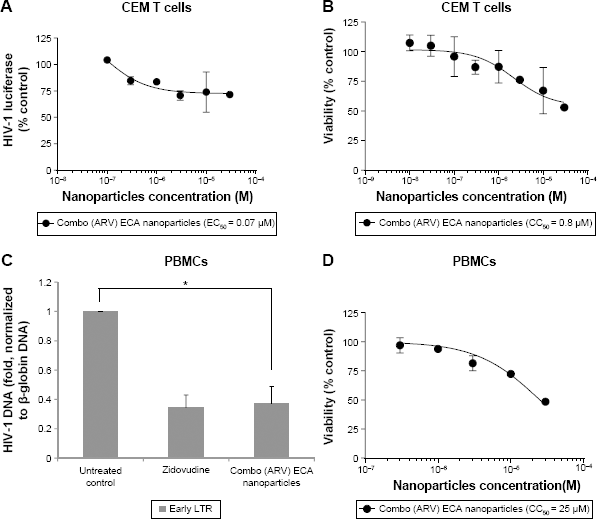
Inhibition of HIV-1 one round infection by ECA nanoparticles. (
Taken together, our results indicate that HIV-1 inhibition can be achieved by PMM- and ECA-based nanoparticles fabricated by dispersion polymerization and loaded with ARV drugs. As expected, one-round HIV-1 replication was inhibited at the level of RT. These nanoparticles, fabricated by dispersion polymerization, are very promising, as shown in our current data and as exemplified by our recent report on the nanoparticle packaging of HIV-1-activating small molecule, SMAPP1, which was not stable in mouse serum; its activity was preserved and the stability was improved when encapsulated in similar nanoparticles. 39 We also have preliminary data showing successful nanoparticle packaging of HIV-1 transcription inhibitor, 1E7-03 (Nekhai and Akala, unpublished observation).
Conclusion
We show here that four different ARV drugs (zidovudine [nucleoside reverse transcriptase inhibitor], lamivudine [nucleoside reverse transcriptase inhibitor], nevirapine [nonnucleoside reverse transcriptase inhibitor], and raltegravir [HIV integrase strand transfer inhibitor]) could be loaded into nanoparticles by dispersion polymerization and released in vitro. Furthermore, we show here that PMM and ECA nanoparticles loaded with the four antiretroviral drugs efficiently inhibited HIV-1 infection in CEM T cells and PBMCs. As expected, ARV-loaded nanoparticles inhibited HIV-1 RT. The ARV-loaded nanoparticles may help to improve the efficacy of ARV drugs. The ARV-loaded nanoparticles with PEG on the corona may facilitate tethering ligands for targeting specific receptors expressed on the cells of HIV reservoirs. Other advantages of the nanoparticles reported here are as follows: the fabrication of the nanoparticles is a one-pot process (simultaneous encapsulation of ARV drugs during polymerization) carried out at ambient temperature; it is possible to add surface functionalities in one-batch process without further modifications compared to nanoparticle fabrication by dispersion of preformed polymers; the process is surfactant free; and the ARV drugs combination is typical of clinical recommendations. We are optimizing these nanoparticles to target them to the gut and the brain. In conclusion, the ARV-loaded nanoparticles will be useful for future ARV therapy.
Author Contributions
Conceived and designed the experiments: EOA, SN, and WAA. Analyzed the data: EOA, SN, WAA, OO, NK, KAS, OB, SA, and AG. Wrote the first draft of the manuscript: EOA, SN, WAA, OO, NK, KAS, OB, SA, and AG. Contributed to the writing of the manuscript: EOA, SN, WAA, OO, NK, KAS, OB, SA, and AG. Agreed with manuscript results and conclusions: EOA, SN, WAA, OO, NK, KAS, OB, SA, and AG. Jointly developed the structure and arguments for the paper: EOA, SN, WAA, OO, NK, KAS, OB, SA, and AG. Made critical revisions and approved the final version: EOA, SN, WAA, OO, NK, KAS, OB, SA, and AG. All the authors reviewed and approved the final manuscript.