
Abstract
Inhibition of soluble matrix metalloproteinase (MMP) activity is among the non-antibiotic cellular effects exerted by the anti-inflammatory tetracycline derivative minocycline. The impact of minocycline on the signal transduction functions of membrane-bound MMPs is however unknown. We assessed minocycline in a concanavalin-A (ConA)-activated human HepG2 hepatoma cell model, a condition known to increase the expression of membrane type-1 MMP (MT-MMP) and to trigger inflammatory and autophagy processes. We found that minocycline inhibited ConA-induced formation of autophagic acidic vacuoles, green fluorescent microtubule-associated protein 1 light chain 3 (GFP-LC3) puncta formation, gene and protein expression of autophagy biomarker BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3), invasion biomarker MT1-MMP, and inflammation biomarker cyclooxygenase (COX)-2. Gene silencing of MT1-MMP abrogated ConA-induced formation of autophagic acidic vacuoles and ConA-induced expressions of BNIP3 and COX-2. Minocycline was also shown to inhibit ConA-induced signal transducer and activator of transcription 3 (STAT3) phosphorylation as well as gene expression of NANOS1, a biomarker believed to colocalize with MT1-MMP and the specific silencing of which further inhibited ConA-induced STAT3 phosphorylation. Collectively, our data demonstrate that part of minocycline's effects on autophagy could be exerted through the inhibition of MT1-MMP signaling functions, which contribute to the autophagy and inflammatory phenotype of ConA-activated HepG2 cells.
Keywords
Introduction
Cancer cells evolve within a stressful pathological setting characterized by lack of nutrients and an inadequate oxygen supply, combined with a proinflammatory microenvironment. 1 Such conditions, in turn, lead to further types of stresses including oxidative stress, which collectively can lead to cell death through autophagy, a homeostatic mechanism that regulates the turnover of long-lived or damaged proteins and organelles. 2 Autophagy is a highly adaptative metabolic process that plays an important role in stressful conditions regulating cancer cells growth and proliferation,3–5 and that adds up to the resisting cell death molecular signature of cancer. 6 Interestingly, hepatomas are characterized by a chronic proinflammatory environment that contributes to tumor progression. In fact, more than 90% of hepatocellular carcinomas develop within the context of chronic liver damage and inflammation. 7 The molecular interplay between inflammatory and autophagic cues in hepatomas is currently poorly understood.
Similar to what is seen with inflammation, it was recently shown that autophagy enhanced hepatocellular carcinoma progression and cell invasion, 8 in part through activation of the epithelial-to-mesenchymal transition (EMT). 9 More importantly, autophagy has been demonstrated to contribute to the survival of CD133+ liver cancer stem cells, 10 and it was inferred that autophagy inhibition may improve anti-cancer treatments. 11 However, the main problem with current conventional chemotherapy treatments is the adaptive response that leads to chemoresistance; through this process, close molecular cross-talks have been identified between autophagy and liver cancer.12,13 In fact, autophagy is strongly present in many types of cancer cells, and this adaptative activity is a major factor related to tumor progression. 5
Moreover, given that autophagy has recently attracted attention with respect to programed cell death, 14 it is reasonable to hypothesize that any approach that would interfere with autophagy in cancer cells could therefore lead to increased cell death and to reduced resistance of cells to a given therapeutic agent.
The tetracycline-derived antibiotic minocycline was recently reported to inhibit inflammatory15,16 and invasive processes,17,18 which contribute to tumor progression. While current evidence already validates minocycline as a potential therapeutic drug for multiple neurodegenerative disorders, including Parkinson's disease and Huntington's disease, 19 its pharmacologic effects on hepatoma cells and on autophagy processes remain unknown. Furthermore, although inhibition of glioma growth by minocycline was recently reported to be mediated through endoplasmic reticulum (ER) stress-induced apoptosis and autophagic cell death, 20 the precise mechanism of action involved remains unknown.
Membrane type-1 matrix metalloproteinase (MT1-MMP) belongs to the MMP family of enzymes that play an important role in extracellular matrix (ECM) degradation, allowing angiogenesis and tumor invasion.21,22 It has been recently demonstrated that MT1-MMP further triggered inflammatory,23–25 autophagic, 26 and ER stress signaling. 27 Accordingly, a role of MT1-MMP as a cell death sensor/effector through the regulation of ER stress was reported in U87 glioblastoma cells. 27 In fact, MT1-MMP was shown to be involved in concanavalin-A (ConA)-induced autophagy as reflected by increased expression of biomarkers such as BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3), to elicit an inflammatory response through MT1-MMP-mediated increases in cyclooxygenase (COX)-2.25,26
Given that ConA highly induces MT1-MMP and that the clinically approved antibiotic minocycline showed promising properties for adjuvant therapy against malignant gliomas, in part through inhibiting microglial MT1-MMP expression, 28 we sought to investigate further the impact of minocycline on MT1-MMP-mediated inflammation and autophagy in a ConA-activated HepG2 hepatoma cell model.
Materials and Methods
Materials
Sodium dodecyl sulfate (SDS), ConA, minocycline, and bovine serum albumin (BSA) were purchased from Sigma (Oakville, ON). Cell culture media were obtained from Life Technologies (Burlington, ON). Electrophoresis reagents were purchased from Bio-Rad (Mississauga, ON). The enhanced chemiluminescence (ECL) reagents were from Amersham Pharmacia Biotech (Baie-D'Urfé, QC). Micro bicinchoninic acid protein assay reagents were from Pierce (Rockford, IL). The anti-STAT3 (79D7) and anti-phospho-STAT3 (Tyr 705) polyclonal antibodies were from Cell Signaling Technology (Beverly, MA). The polyclonal antibodies against the MT1-MMP catalytic domain and against BNIP3 were from Chemicon (Temecula, CA). Monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was from Advanced Immunochemical (Long Beach, CA). The polyclonal antibody against COX-2 was from Cayman Chemical (Ann Arbor, MI).
Cell cultures
Human hepatocellular HepG2 carcinoma cells were purchased from American Type Culture Collection (ATCC; HB-8065, Manassas, VA). Serum starvation was performed by culturing the cells in Eagle's minimal essential medium (EMEM; GibcoBRL) with 100 units/mL Penicillin/Streptomycin, from which the 10% inactivated fetal bovine serum (Hyclone Laboratories, Logan, UT) was omitted.
Detection of autophagic acidic vesicular organelles
HepG2 cells were serum-starved following the different conditions of treatment with ConA or minocycline, or following siRNA transfections. Acridine Orange (0.5 μg/mL; Sigma-Aldrich Canada, A6014) was added to each well, and cells were incubated for 10 minutes at 37 °C in the dark. Fluorescence was then examined by microscopy using a Nikon Eclipse TE2000-U microscope. Micro-Manager 1.3.39 imaging software (Copyright University of California, San Francisco, 2007) was used to capture images with a QImaging Retiga 1300 camera (Nikon), and data were analyzed with ImageJ MacBiophotonics (National Institutes of Health (NIH)). Representative images display the red channel (568 nm), which were then extracted as a grayscale image using ImageJ. Mean fluorescence was quantified as the following ratio: (total red fluorescence of sample–-total red fluorescence of sample background)/(total red fluorescence of control–-total red fluorescence of control background).
Detection of green fluorescent microtubule-associated protein 1 light chain 3 (GFP-LC3) puncta formation
HepG2 cells were harvested on cover slips, transiently transfected with a cDNA plasmid encoding pEGFP-LC3 (generously provided by Dr Patrick Labonte, INRS-IAF, QC). Upon ConA treatment, medium was removed and cells were fixed in 10% formalin phosphate buffer (Fisher Scientific, Ottawa, ON) for 20 minutes, and then blocked for one hour in 1% BSA/PBS/NaN3. A solution of 10 μg/mL 4′, 6′-diamidino-2-phénylindol diluted in PBS was used to stain the nuclei. Puncta formation was then examined in 5–10 transfected cells and quantified by fluorescent microscopy using a Nikon Eclipse TE2000–U microscope. The Micro-Manager 1.3.39 imaging software was then used to capture images with a QImaging Retiga 1300 camera, and data were analyzed with ImageJ Macbiophotonics (NIH). Representative images displaying the GFP channel (488 nm) were then extracted as a grayscale image using ImageJ. GFP-LC3 puncta formation was defined as bright dots ≫1.5 SD above the mean cytosolic fluorescence.
Immunoblotting procedures
HepG2 cells were lysed, and proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE). After electrophoresis, proteins were electrotransferred to polyvinylidene difluoride membranes, and immunoreactive material was visualized by ECL. 29
Total RNA isolation, CDNA synthesis, and realtime quantitative RT-PCR
Total RNA was extracted from HepG2 monolayers using TRIzol reagent (Life Technologies, Gaithersburg, MD). For cDNA synthesis, 1 μg of total RNA was reverse-transcribed into cDNA using a high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). cDNA was stored at −80 °C before PCR. Gene expression was quantified by real-time quantitative PCR using iQSYBR Green Supermix (Bio-Rad, Hercules, CA). DNA amplification was carried out using iCycler iQ5 (Bio-Rad, Hercules, CA), and product detection was performed by measuring binding of the fluorescent dye SYBR Green I to double-stranded DNA. The following primer sets were provided by QIAGEN (Valencia, CA): MT1-MMP (HS_Mmp14_1_SG QT00001533), BNIP3 (HS_BNIP3_1_SG QT00024178), COX-2 (HS_PTGS2_1_SG QT00040586), and NANOS1 (HS_ NANOS1_1_SG QT00219541). The relative quantities of target gene mRNA against an internal control (β-Actin) RNA were measured by following a δCT method employing an amplification plot (fluorescence signal vs. cycle number). The difference (δCT) between the mean values in the triplicate samples of target gene and those of β-Actin RNA was calculated by CFX manager software version 2.1 (Bio-Rad), and the relative quantified value (RQV) was expressed as 2−ΔCT.
Transfection method and RNA interference
HepG2 cells were transiently transfected with 20 nM siRNA against STAT3 (HS_Stat3_7 FlexiTube siRNA, S102662338), MT1-MMP (HS_Mmp14_6 HP siRNA, S103648841), NANOS1 (HS_NANOS1_5, S104177733), or scrambled sequences (AllStar Negative Control siRNA, 1027281) using the DharmaFECT transfection reagent (Thermo Fisher Scientific, Waltham, MA). The small interfering RNA and mismatch siRNA were all synthesized by QIAGEN and annealed to form duplexes.
Statistical data analysis
Data are representative of three or more independent experiments. Statistical significance was assessed using Student's unpaired t-test. Probability values of less than 0.05 were considered significant, and an asterisk denotes such significance in the figures.
Results
Minocycline abrogates autophagic acidic vacuole formation, GFP-LC3 puncta formation, and expression of BNIP3 in ConA-activated HepG2 cells
ConA is a lectin reported to trigger inflammation and ECM degradation through MT1-MMP-mediated processes.24,25,30 More relevantly, ConA was recently found to trigger autophagy in U87 glioblastoma cells, a process known to involve MT1-MMP. 26 We therefore tested whether minocycline affected ConA-induced autophagy in HepG2 cells. Serum-starved HepG2 cells were treated with 30 μg/mL ConA in the presence of various concentrations of minocycline. Acridine Orange staining was then used to assess the extent of autophagic acidic vacuole formation (Fig. 1A, upper panels) as well as GFP-LC3 puncta formation (Fig. 1A, lower panels) whose assays are indicative of autophagy processes. While both the Acridine Orange staining and GFP-LC3 signal in response to 10 μM minocycline were absent (not shown), we found that ConA-induced acidic vacuole and GFP-LC3 puncta formation (Fig. 1B) and that minocycline dose-dependently antagonized that induction with a maximal effect at 3 μM (Fig. 1C). To further document the molecular effects of minocycline on ConA-induced autophagy, we used immunofluorescent staining to assess the intracellular expression of autophagy biomarker BNIP3 (Fig. 2A). We found that cytoplasmic BNIP3 expression was significantly increased in ConA-activated HepG2 cells and that minocycline completely abolished that increase (Fig. 2B).

Minocycline inhibits the induction of acidic vacuoles in ConA-activated HepG2 cells. Serum-starved HepG2 hepatoma cells were treated with or without 30 μg/mL ConA for 24 hours in the presence of various concentrations of minocycline (0–10 μM). (A) Upper panels: Cells were stained with Acridine Orange as described in the Methods section, and acidic vacuole formation was examined by fluorescent microscopy. Lower panels: Before ConA treatment, cells were transiently transfected with a plasmid cDNA encoding GFP-LC3. Fluorescent puncta formation was assessed as described in the Methods section. (B) ConA-induced autophagy was monitored by Acridine Orange staining (black bars) or upon GFP-LC3 puncta formation (gray bars), and compared to their respective untreated control cells (white bars). (C) Minocycline effect on ConA-treated cells was similarly quantified for different concentrations in ConA-treated cells by Acridine Orange staining (closed circles) or GFP-LC3 puncta formation (gray circles). Acridine Orange staining and GFP-LC3 puncta formation pictures are representative of three independent experiments, and corresponding fluorescence analyses are shown. Statistical significance was assessed using Student's unpaired t-test. Probability values of less than 0.05 were considered significant, and an asterisk (*) denotes such significance.

Minocycline inhibits the induction of BNIP3 in ConA-activated HepG2 cells. Serum-starved HepG2 hepatoma cells were treated with or without 30 μg/mL ConA for 24 hours in the presence of 10 μM minocycline. (A) Cells were fixed and immunostained with either IgG or anti-BNIP3 antibody as described in the Methods section. DAPI-stained nucleus was visualized in blue. (B) Fluorescence quantification was performed in control (white bar) and ConA-treated (black bar) cells and is representative of three independent experiments. Statistical significance was assessed using Student's unpaired t-test. Probability values of less than 0.05 were considered significant, and an asterisk (*) denotes such significance.
Minocycline antagonizes the expression of MT1-MMP and COX-2 proteins in ConA-activated HepG2 cells
In addition to BNIP3 expression, we next determined whether minocycline also altered the expression of MT1-MMP and COX-2, two inter-related inflammation-associated biomarkers well documented to be increased by ConA.24,25 Cell lysates were isolated from ConA-treated HepG2 cells, and we found that ConA dose-dependently triggered MT1-MMP, COX-2, and BNIP3 protein expression (Fig. 3A and B). When minocycline was concomitantly added to cells with ConA, we observed a dose-dependent reversal of the ConA-mediated effects on all three induced biomarkers (Fig. 3C and D). Altogether, this cellular and molecular evidence confirmed that minocycline can reverse the effects of ConA on markers of autophagy and inflammation processes.
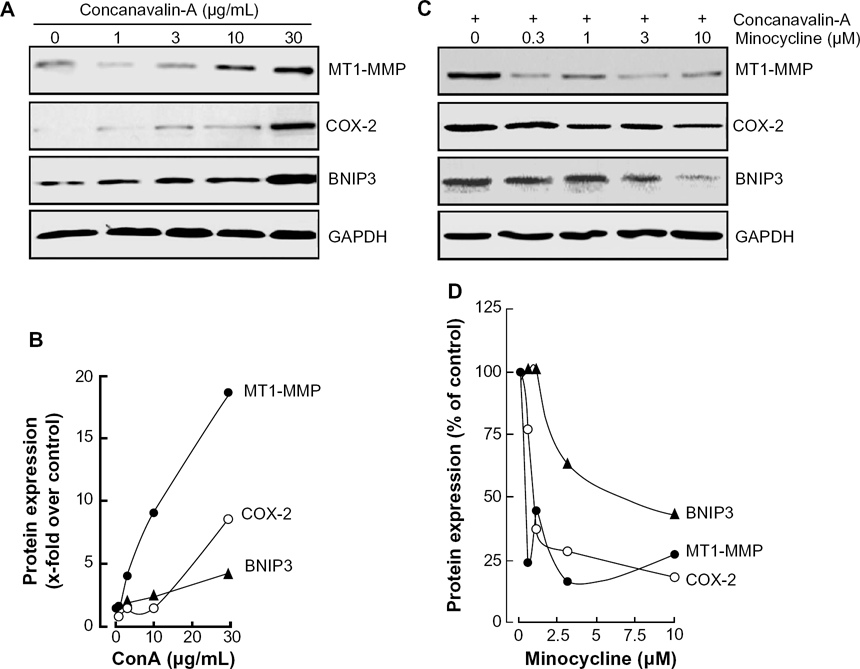
Minocycline antagonizes the protein expression of BNIP3, MT1-MMP, and COX-2 in ConA-activated HepG2 cells. Serum-starved HepG2 hepatoma cells were treated with various ConA concentrations for 24 hours (A), or co-treated with ConA (30 μg/mL) having various concentrations of minocycline (C). Cell lysates were isolated and electrophoresed as described in the Methods sections. Scanning densitometry measurements were performed (B, D) on two independent experiments. A representative scanning densitometry profile is shown for the respective treatments. Data represent mean values in duplicates.
MT1-MMP is required in ConA-induced BNIP3 expression and formation of autophagic acidic vacuoles
Given that MT1-MMP was recently ascribed intracellular signal transducing functions in inflammation and in autophagy,24,26 we next assessed the contribution of MT1-MMP to ConA-mediated induction of BNIP3, COX-2, and autophagic acidic vacuole formation in HepG2 cells. Specific MT1-MMP gene silencing was performed as described in the Methods section and was shown to antagonize ConA-induced BNIP3 and COX-2 protein expression as efficiently as does minocycline (Fig. 4A). Gene expression was also assessed under the same conditions, and similar effects were observed for MT1-MMP, BNIP3, and COX-2 transcriptional regulation (Fig. 4B), suggesting that ConA requires MT1-MMP to relay intracellular signaling that affects gene transcription.
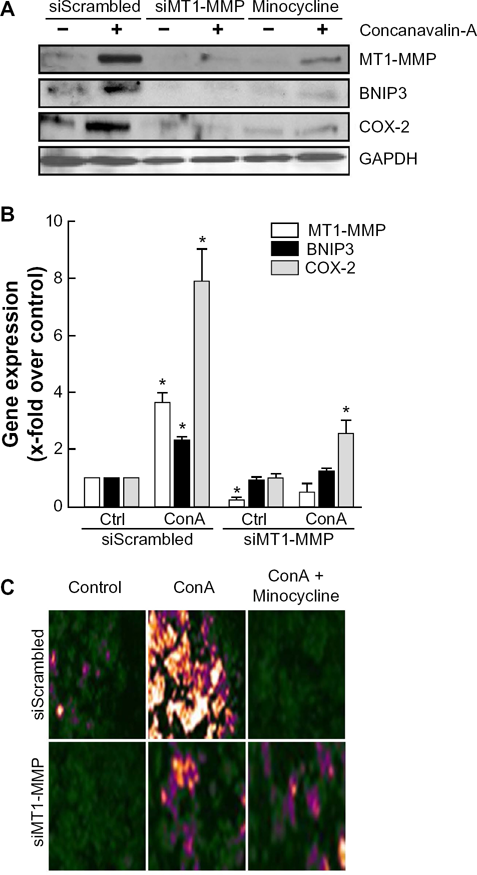
MT1-MMP is required for ConA-induced BNIP3 expression and formation of acidic vacuoles. Serum-starved HepG2 hepatoma cells were transiently transfected with a control siRNA (siScrambled) or an siRNA directed against MT1-MMP (siMT1-MMP); then the cells were treated with 30 μg/mL ConA for 24 hours in the presence or absence of 10 μM minocycline. (A) Cell lysates were isolated and electrophoresed, and immunodetection performed as described in the Methods section. (B) Total RNA was isolated from cells transfected with either a scrambled siRNA sequence (siScr) or a specific siRNA against MT1-MMP (siMT1-MMP), and followed by 30 μg/mL ConA treatment. cDNA was then synthesized and qRT-PCR performed as described in the Methods section. A representative qPCR profile, out of three independent experiments, is shown for the corresponding genes. Data represent mean values in triplicates. Statistical significance was assessed using Student's unpaired t-test. Probability values of less than 0.05 were considered significant, and an asterisk (*) denotes such significance. (C) Cells were stained with Acridine Orange as described in the Methods section, and acidic vacuole formation was examined by fluorescence microscopy in control, ConA-, and ConA/minocycline-treated cells.
When autophagic acidic vacuole formation was assessed, we observed that ConA triggered autophagy and that this was similarly antagonized upon either MT1-MMP silencing or minocycline treatment (Fig. 4C). Altogether, these data suggest that MT1-MMP-mediated signaling is mandatory in the induction of autophagy and inflammation in ConA-activated HepG2 cells and that minocycline may efficiently affect these processes by regulating MT1-MMP expression itself.
STAT3 and NANOS1 are important signaling and upstream regulators in ConA-induced MT1-MMP
Recent evidences has indicated a role of STAT3-mediated signaling in the transcriptional regulation of COX-2 and of colony-stimulating factors in ConA-activated mesenchymal stromal cells.24,29 Given that NANOS1 has also been suggested to upregulate MT1-MMP expression at both the gene and protein levels in epithelial tumor cells, 31 we therefore decided to silence STAT3 and NANOS1 gene expressions and assess their respective impact on ConA-induced MT1-MMP expression. We found that the induction of MT1-MMP protein expression by ConA was abrogated in conditions where either STAT3 or NANOS1 was silenced (Fig. 5A). When MT1-MMP gene expression was assessed, we further found that minocycline significantly reduced the basal levels of MT1-MMP and that this inhibitory effect required the expression of STAT3 and NANOS1 (Fig. 5B, black bars). Interestingly, NANOS1 silencing also abrogated ConA-induced gene expression of MT1-MMP whereas STAT3 silencing still allowed weak induction of MT1-MMP by ConA (Fig. 5B, gray bars). STAT3 and NANOS1 therefore represent crucial upstream contributors in the transcriptional regulation of MT1-MMP by ConA, and in the mechanism of action of minocycline. Finally, we also show that specific gene silencing of NANOS1 and of STAT3 was achieved and did not affect the mRNA levels of each other (Fig. 5C).
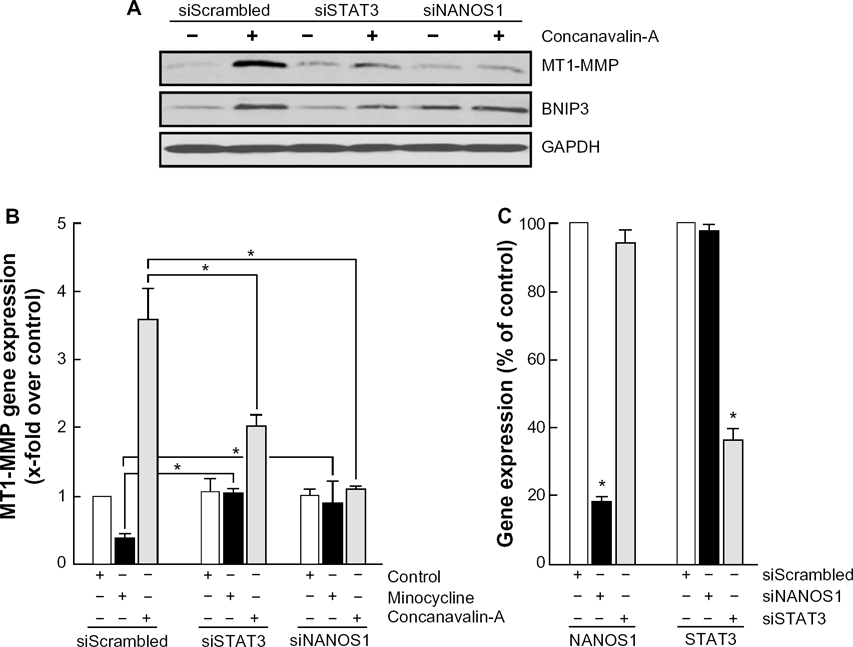
STAT3 and NANOS1 gene silencing abrogate ConA-induced MT1-MMP and BNIP3 expression. HepG2 hepatoma cells were transiently transfected with a control siRNA (siScrambled) or an siRNA directed against STAT3 (siSTAT3) or NANOS1 (siNANOS1); then serum-starved cells were treated with 30 μg/mL ConA for 24 hours. (A) Cell lysates were isolated and electrophoresed, and immunodetection performed as described in the Methods sections. (B) MT1-MMP gene expression was assessed in cells that were treated with 10 μM minocycline or 30 μg/mL ConA. Total RNA was isolated, cDNA synthesized, and qRT-PCR performed as described in the Methods section. (C) The inter-relationship between NANOS1 and STAT3 gene expression was assessed in cells that were either silenced for NANOS1 (siNANOS1, black bars) or silenced for STAT3 (siSTAT3, gray bars). Total RNA was isolated, cDNA synthesized, and qRT-PCR performed as described in the Methods section. A representative qPCR profile, out of three independent experiments, is shown for the corresponding genes. Data represent mean values in triplicates. Statistical significance was assessed using Student's unpaired t-test. Probability values of less than 0.05 were considered significant, and an asterisk (*) denotes such significance.
Minocycline inhibits ConA-induced NANOS1 gene expression and phosphorylation of STAT3
Given the molecular rationale that links NANOS1 to MT1-MMP, we next tested their interdependence regarding ConA action. HepG2 cells were treated with various concentrations of ConA, which resulted in a linear correlation (r2 = 0.976) between NANOS1 and MT1-MMP expression, and which confirmed that ConA triggers MT1-MMP and NANOS1 in a dose-dependent manner (Fig. 6A). Minocycline, which had no effect on basal endogenous NANOS1 expression (not shown), was next added to ConA-treated cells and was found to decrease both MT1-MMP and NANOS1 gene expression levels in a dose-dependent fashion (Fig. 6B). The impact of NANOS1 was also assessed in ConA signaling. We found that ConA effectively triggered STAT3 phosphorylation over time, and that minocycline lowered this phosphorylation (Fig. 6C). When NANOS1 gene expression was silenced, ConA was unable to trigger STAT3 phosphorylation (Fig. 6D). Collectively, this suggests that NANOS1 is an important upstream intermediate in the ConA-mediated signaling, which can also be targeted by minocycline.
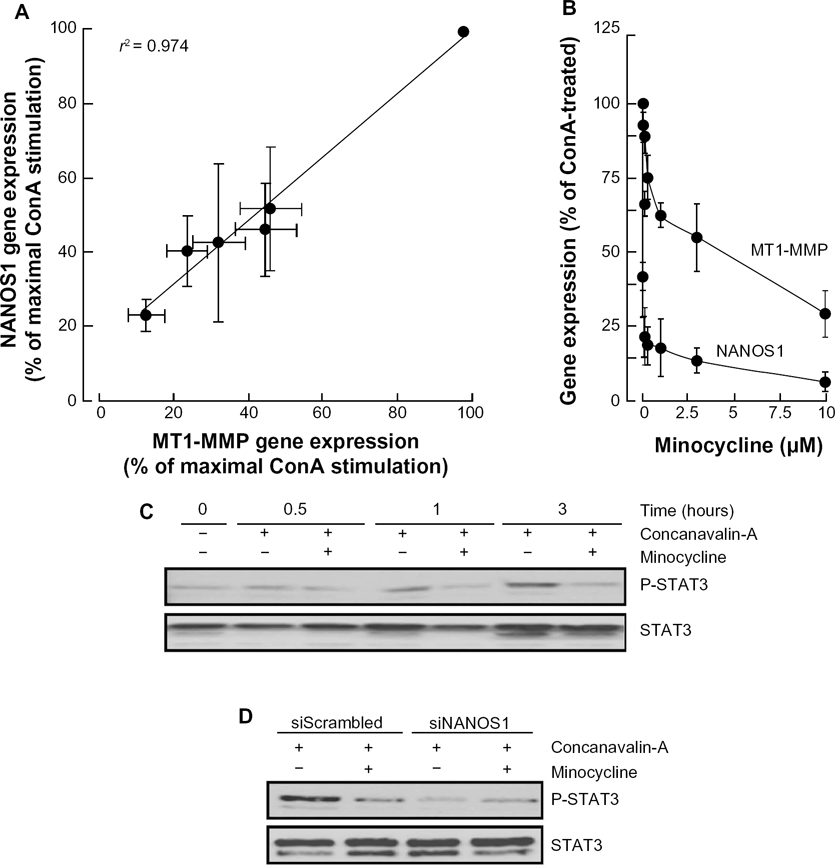
Minocycline inhibits ConA-induced NANOS1 expression and phosphorylation of STAT3. (A) Serum-starved HepG2 hepatoma cells were treated with increasing ConA concentrations. NANOS1 and MT1-MMP gene expression was expressed as the percentage of maximal ConA-induced effect for each gene. (B) ConA-treated HepG2 hepatoma cells were incubated with up to 10 μM minocycline for 24 hours. Total RNA was isolated, cDNA synthesized, and qPCR performed as described in the Methods section. The levels of STAT3 phosphorylation were assessed in cells that were (C) treated with 30 μg/mL ConA in the presence or absence of 10 μM minocycline or (D) transiently transfected with either a control siRNA (siScrambled) or an siRNA directed against NANOS1 (siNANOS1). A representative qPCR profile, from two independent experiments, is shown for the corresponding genes. Data represent mean values from triplicates.
ConA-induced autophagic acidic vacuole formation requires NANOS1 and STAT3
Our previous data showed the absolute requirement for MT1-MMP in ConA-induced autophagic acidic vacuole formation (Fig. 4C). We next examined the involvement of NANOS1 and of STAT3 in the ConA-mediated induction of acidic vacuoles. Transient gene silencing was performed for NANOS1, STAT3, and MT1-MMP, and then cells were treated with ConA and stained with Acridine Orange. We observed diminished acid vacuole formation in both MT1-MMP and STAT3 silencing, while incomplete inhibition was observed in cells silenced for NANOS1 gene expression (Fig. 7).

ConA-induced autophagic acidic vacuole formation requires NANOS1 and STAT3. HepG2 hepatoma cells were transiently transfected with a control siRNA (siScrambled) or an siRNA directed against MT1-MMP (siMT1-MMP), STAT3 (siSTAT3), or NANOS1 (siNANOS1). Serumstarved cells were treated with 30 μg/mL ConA for 24 hours, and then cells were stained with Acridine Orange as described in the Methods section, and acidic vacuole formation was examined by fluorescence microscopy.
Discussion
Recently reported MT1-MMP-associated intracellular signaling functions link MT1-MMP-mediated ER stress, autophagy, and inflammation to the invasive and chemoresistance signature of cancer cells.5,6 In line with these new functions, the present study further strengthens the conception that ConA-induced autophagy and inflammation processes require a combined MT1-MMP upstream induction and MT1-MMP downstream signaling pathways’ activation, which involves NANOS1 and which can both be abrogated by minocycline.
Our finding that minocycline efficiently alters the relationship that exists between MT1-MMP and NANOS1 expression (Fig. 6A) does not only impact on inflammation/autophagy processes but also on the acquisition of chemoresistance and invasive properties of cancer cells, often correlated to the EMT process. Indeed, NANOS1 is a protein involved in a variety of processes including cell migration and oogenesis in Drosophila.32,33 NANOS1 expression is tightly controlled by the cell surface adhesion molecule E-cadherin, the lowered expression of which is frequently associated with tumor formation and progression. 34 In fact, high expression of NANOS1 was reported to correlate with low levels of E-cadherin and with aggressive tumor behavior.35,36 Interestingly, overexpression of MT1-MMP was recently demonstrated to induce both EMT processes associated with breast tumor progression 37 and the generation of cells exhibiting cancer stem cells properties and decreased expression of E-cadherin. 38 How NANOS1 induces invasiveness still remains unclear but has been, in part, associated with MT1-MMP upregulation. 31 In fact, NANOS1 overexpression upregulated MT1-MMP at the mRNA and protein levels, and this was shown to promote cell migration. 31 It is however still unclear whether the transducing events involved in this process may also concomitantly trigger some autophagy. Our data however confirm NANOS1 involvement in ConA-induced MT1-MMP and BNIP3 expression and autophagic acidic vacuole formation. Given that the interdependency between NANOS1 and MT1-MMP decreases upon minocycline treatment, it is tempting to hypothesize that EMT processes can also be efficiently targeted in this way.
A new finding that we bring to our understanding of ConA-induced MT1-MMP mechanism is the role of STAT3 and more specifically of its phosphorylated intermediate.
Silencing of STAT3 leads to a reduction in ConA-induced MT1-MMP, while NANOS1 gene silencing also abrogated this phosphorylation event. Whether the effect of NANOS1 knockdown on STAT3 phosphorylation is a secondary effect because of inflammation is indirectly supported by the fact that minocycline efficiently reversed ConA-induced COX-2 expression (Fig. 4A). However, this remains to be further investigated possibly through gene silencing strategies of COX-2. Given that minocycline efficiently inhibited ConA-induced NANOS1 expression, ConA-induced STAT3 phosphorylation, and ConA-induced autophagic acidic vacuole formation, we suggest that ConA-mediated signaling can trigger a pathway involving the NANOS1/pSTAT3 signaling axis to induce MT1-MMP (Fig. 8). These observations indicate a potential dual role for pSTAT3 that would take place not only upstream of MT1-MMP induction through NANOS1 but also downstream. Indeed, once induced, MT1-MMP triggers inflammation and autophagy through pathways that involve STAT3 phosphorylation. It was shown that the ConA-induced inflammatory response is mediated by MT1-MMP induction, which, in turn, activates the JAK/STAT3 signaling pathway in mesenchymal stromal cells. 24
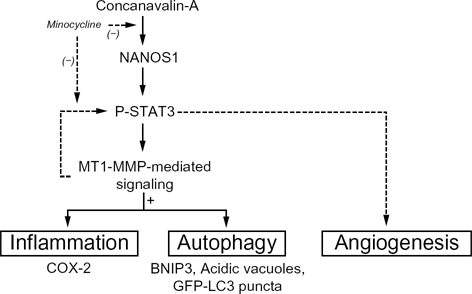
Minocycline exerts anti-inflammatory and anti-autophagy action in ConA-activated hepatoma cells. ConA triggers STAT3 phosphorylation and NANOS1 gene expression in HepG2 cells. NANOS1 and STAT3 phosphorylation is upstream prerequisite for ConA-mediated MT1-MMP gene expression, which, in turn, triggers the inflammatory biomarker COX-2 expression, as well as the autophagy biomarker BNIP3 and acidic vacuole formation. Minocycline can exert its effects by inhibiting ConA-induced STAT3 phosphorylation, or directly on either MT1-MMP or NANOS1 transcriptional regulation. Dotted lines: Minocycline exerts anti-angiogenic effects. 47 An alternate feedback loop was recently suggested to occur, 39 where MT1-MMP contributed to STAT3 phosphorylation, which in turn impacted on proangiogenic transcription of proangiogenic cytokines.
More recently, ConA-induced MT1-MMP, possibly through toll-like receptor (TLR) activation, also triggered JAK/STAT3 signaling to generate proangiogenic and immunomodulatory cytokine expression (Fig. 8, dotted lines).39,40 Interestingly, recent observations point to a potential anti-angiogenic property of minocycline. 41 Whether minocycline also inhibits MT1-MMP-mediated angiogenesis is currently unknown. On the other hand, what is known is that STAT3 gene silencing does not prevent ConA-induced proMMP-2 activation, an event that requires MT1-MMP induction and targeting at the cell surface. 24 It is therefore unlikely that STAT3 phosphorylation status may modulate MT1-MMP expression. How ConA may trigger NANOS1 transcription remains unknown to this date. Several transduction pathways have been recently reported to be triggered by ConA and these included ERK, RhoA/ROK, JAK/STAT, and NFκB. Unfortunately, the exact mechanisms involved remain elusive although evidence suggests that cell surface TLR may contribute to transduce ConA signaling.39 As for minocycline's anti-ConA effects observed in this study, the only conclusions that can be safely withdrawn are that it acts as a signal transducer inhibitor affecting gene transcription that will be the object of future investigations.
In addition to its antibiotic and anti-inflammatory properties, minocycline could therefore be envisioned as a promising agent to complement current cancer treatment modalities because of its combined anti-inflammatory and anti-tumoral effects. Indeed, it has been shown that minocycline inhibited growth of several types of tumor cells including epithelial ovarian cancer cells and glioma cells.20,42 Interestingly, doxocycline is among other members of the tetracycline molecules which, similar to minocycline, possesses MMP inhibitory functions. Although doxocycline's mechanism of action on cancer cell growth remains unclear, it also has been associated with suppression of MMP expression and, more specifically of MT1-MMP, in pre- clinical models of malignant epithelial cells. 43 Whether similar inhibitory mechanisms were also involved in doxocycline effects on melanoma, 44 renal and prostate cancer, 45 and breast cancer 46 remains to be confirmed. Accordingly, in this study we demonstrate that minocycline inhibits ConA-induced MT1-MMP in a HepG2 hepatoma cell model.
Minocycline's anti-inflammatory properties are also believed to be mediated through the inhibition of proinflammatory cytokines such as TNF-α and IL-6.18,41 We further demonstrated here that inflammation, as monitored by COX-2 induction, requires an MT1-MMP/STAT3 signaling axis 24 and is inhibited by minocycline in ConA-activated HepG2 cells. Altogether, these observations suggest that minocycline could alter the MT1-MMP-mediated JAK/STAT3 signaling axis, which makes kinases upstream from the JAK family possible targets for minocycline. We also present evidence that ConA-induced autophagy is inhibited by minocycline as shown by decreased BNIP3, GFP-LC3 puncta formation, and autophagic acidic vacuole formation. As minocycline also inhibits MT1-MMP expression, one could hypothesize that minocycline abrogates ConA-induced autophagy by preventing MT1-MMP expression and/or signaling functions. Given that ConA was reported to activate and transduce part of its signaling through cell surface TLR receptors, 39 minocycline may therefore also be envisioned as targeting some TLR receptor-mediated signaling.
We conclude that in HepG2, ConA-induced inflammation and autophagy are mediated through MT1-MMP and can be inhibited by minocycline. We suggest that minocycline exerts part of its non-antibiotic functions not only through the inhibition of inflammation and autophagy by targeting signaling intermediates involved in upstream regulation of MT1-MMP such as TLR or NANOS1 but also by impairing subsequent downstream signaling pathways such as JAK/STAT3.
Footnotes
Acknowledgments
BA holds a Canada Research Chair in Molecular Oncology from the Canadian Institutes of Health Research (CIHR). JP is a Natural Sciences and Engineering Research Council of Canada (NSERC) awardee.
Author Contributions
Conceived and designed the experiments: MD and BA. Performed the experiments: MD, JP, AL. Analyzed the data: MD, JP, AL, CM, KH, and BA. Wrote the first draft of the manuscript: MD, CM, KH, and BA. Contributed to the writing of the manuscript: JP and AL. Agree with manuscript results and conclusions, made critical revisions and approved the final version of manuscript: MD, JP, AL, CM, KH, and BA. All authors reviewed and approved of the final manuscript.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.
Abbreviations
BNIP3, BCL2/adenovirus E1B 19 kDa interacting protein 3; ConA, concanavalin-A; COX, cyclooxygenase; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; ER, endoplasmic reticulum; MMP, matrix metalloproteinase; GFP-LC3, green fluorescent microtubule-associated protein 1 light chain 3; MT1-MMP, membrane type-1 MMP; STAT, signal transducer and activator of transcription; TLR, toll-like receptor.