
Abstract
With the increasing number of sequenced genomes and their comparisons, the detection of orthologs is crucial for reliable functional annotation and evolutionary analyses of genes and species. Yet, the dynamic remodeling of genome content through gain, loss, transfer of genes, and segmental and whole-genome duplication hinders reliable orthology detection. Moreover, the lack of direct functional evidence and the questionable quality of some available genome sequences and annotations present additional difficulties to assess orthology. This article reviews the existing computational methods and their potential accuracy in the high-throughput era of genome sequencing and anticipates open questions in terms of methodology, reliability, and computation. Appropriate taxon sampling together with combination of methods based on similarity, phylogeny, synteny, and evolutionary knowledge that may help detecting speciation events appears to be the most accurate strategy. This review also raises perspectives on the potential determination of orthology throughout the whole species phylogeny.
Keywords
Introduction
Detection of orthologs is of fundamental importance in many fields of biology, particularly in the annotation of newly sequenced organisms, functional genomics, gene organization in species, evolutionary studies of biological systems, and phylogenomic analyses. Accurate determination of evolutionary relationships between gene (protein) families involving multiple species is of utmost importance for such goals.
Starting in 1995 with the first complete genome sequence of a free-living organism, Haemophilus influenzae, 1 genomics has opened a new research era on species evolution. The availability of full genome datasets raised the hope to decipher species relationships in terms of evolution. Only a few years later, the accumulation of complete genome sequences from three phylogenetic domains has revealed the existence of significant evolutionary processes such as gene exchange between species,2,3 partial and whole-genome duplication, 4 and gene loss,5,6 casting doubt on the established species tree topology. Moreover, significant incongruence was observed between species and gene tree topologies, especially in bacteria where horizontal gene transfers (HGTs) tend to blur the phylogeny of species7–9 and, therefore, the accurate detection of orthologs. The tree representation of cell life was thus questioned, and its replacement by a net or ring of life7,10 suggested to reflect gene exchange between species. With the exponential increase of available full genome sequences and their studies, 11 several new methods that take into account these evolutionary processes have been introduced for gene and species tree construction.9,12–14
Most methods for large-scale detection of orthologs are based on homology as inferred by sequence similarity. However, proper identification of orthologous genes is a major challenge because the accumulation of evolutionary dynamic events tends to blur the recognition of true orthologs among homologs.15–18 Numerous methods were elaborated to solve this problem, with various advantages and limitations.17,19,20 In general, most of these methods suffer from a common limitation, namely, the difficulty in constructing orthologous classes in the presence of paralogs. For distantly related species, assessing orthology can become quite difficult, typically due to low similarity between protein sequences and the likely increase of gene birth and death. 15 In contrast to closely related species, synteny conservation provides useful information to identify conserved chromosomal segments, in which orthologous genes can be more steadily searched for.
In this review, a reminder of basic definitions concerning homology, paralogy, and orthology relationships is first proposed. Then, some of the numerous computational methods designed to infer orthologs are introduced, along with a discussion on their advantages and limitations. Accurate construction of species trees is directly linked to inference of orthologs. Difficulties related to species tree constructions undermine orthology inference and inversely. Some of these difficulties will be discussed. Finally, some open questions about ongoing efforts in computational development for the detection of orthologous genes are raised.
Concepts of Homology, Orthology, and Paralogy
Figure 1 illustrates the concepts of homology, paralogy, and orthology.
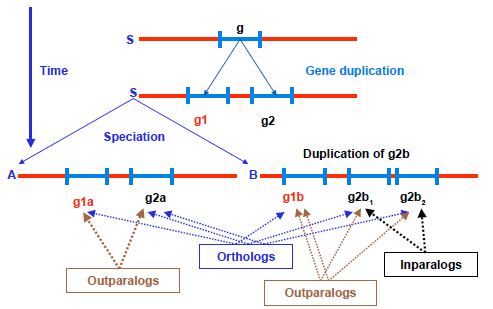
Homologs–-paralogs–-orthologs.
Homology
Homology is a basic concept at the core of evolutionary genomics. Identifying homology relationships between sequences is the first fundamental step in many biological research domains, and more particularly so in inferring orthologs and paralogs. According to Fitch,21,22 two genes are homologs if they share a common origin, ie, derived from a common ancestor. Sequence similarity 23 is almost the only criterion available to infer homology. This criterion introduces a limitation to the detection of homology, because sequences may diverge beyond statistical recognition as the evolutionary distances between species increase. Additional complications include the dynamics of gene duplication, loss, transfer or fusion/fission, and shuffling that occurred during genome evolution.
Orthologs–-paralogs
Orthologs are homologous genes resulting from a speciation event, whereas paralogs are homologous genes resulting from a duplication event. The dynamic of duplication/loss during evolution is such that paralogy does not require paralogous genes to be in the same genome. 16 Depending upon taxon sampling, genes in single copies in two distinct genomes may result from duplicated copies in their ancestor after differential gene loss in the two derived lineages (Fig. 1). Hence, there is a distinction between in-paralogs, corresponding to paralogs issued from duplication post speciation, and out-paralogs, corresponding to duplication prior to speciation. 24 Out-paralogs are also sometimes referred to as pseudoorthologs 16 in configuration with differential gene loss where they appear to be orthologs, while two in-paralogs issued from duplication in one lineage are themselves co-orthologs to the only copy present in another lineage in which no duplication took place.
Specific situations are generated by events of whole-genome duplication (WGD) and by HGT. The numerous pairs of in-paralogs left after a WGD have been designated ohnologs, a term that helps distinguish them from other paralogs resulting from other ancestral duplication. 25 Xenologous genes are genes that appear falsely as orthologs because at least one of the pair is acquired via HGT from another species. Usually it is difficult to identify xenologs in pairwise genome comparisons. Xenologs and true orthologs might be distinguished in multiple genome comparisons if the origin of each gene can be identified. 16
As originally recognized by Ohno, 26 gene duplication creates a novel evolutionary paradigm as the selective functional pressures act at different regimes when genes are single or multiple copy. Thus, paralogous genes tend to rapidly diverge in function.
Evolutionary Processes and Consequences on Ortholog Inference
Large-scale genome comparisons showed that genes and genomes are subject to strong evolutionary dynamics. These evolutionary processes (Fig. 2) include HGT between species, 2 gene loss and acquisition, 27 protein domain emergence, gain and loss28,29 events, and, at the genome level, partial/whole-genome duplications and introgression events. 30 WGD events can take place in one round, as in yeast, 4 or in multiple rounds, as in plants, fishes, and other vertebrates. 31 Additional complications might arise by loss of genes in some descent species obtained after rounds of speciation and WGD events. All these events, together with gene transfer, accumulation, and loss, tend to blur the recognition of true orthologs among a set of homologs.15–18,32
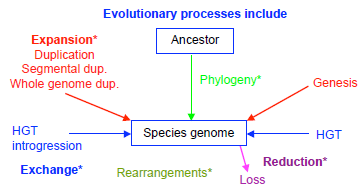
Evolutionary processes.
Horizontal gene transfer–-introgression events
HGT (also called lateral gene transfer) is the transmission of genes from a species to another one through processes distinct from ancestral inheritance (or vertical transfer). HGT has emerged as a major evolutionary process that has shaped genomes in all three domains of life. It has been recognized as one of the major evolutionary forces driving prokaryote evolution. 33 Recent estimates by Dagan et al suggest that on average 81% of prokaryote gene have been involved in HGT at some point in their history. 34 In eukaryotes, HGT occurs on a previously unsuspected scale.35,36
Introgression is another process of lateral transfer that concerns the transmission of regions of a species' genome to the genome of another species, occurring within closely as well as distantly related species.18,37,38
As a result, HGT and introgression events imply different evolutionary histories in the content of the host species, 39 affecting the concepts of evolutionary relationships between species, 40 and hence the detection of paralogs and orthologs and also phylogenetic tree constructions.
Protein domain emergence and gain and loss events
A domain is a structural constituent formed by a distinct region in a specific protein. A domain in a protein sequence might be unique or associated with other domains. Domains are evolutionarily well conserved across taxa 41 and are frequently rearranged (due to duplication, fusion, fission, as well as terminal domain loss) between and within proteins and genomes. 42 Emerging domains (ie, previously unreported) are more likely disordered in structure and spread more rapidly within their genomes than established domains. 29 A significant number of domains are lost along every lineage, 43 and insertions or deletions are more common than substitutions of domains. Domain insertions are significantly more common than domain deletions. 42 In the protein universe, the growth of a single-domain architecture is slow, whereas the growth of a multidomain architecture results from the combination of a single-domain architecture. 44
Protein domains may give rise to supplementary difficulties in orthology inference due to the possible different ancestral origins of the corresponding gene(s) particularly when resulting from fission or fusion events. Different domains may have different ancestors. Proteins with multiple domains may have multiple significant hits with different proteins (each hit might be based on a given domain in the query protein sequence) with different relative positions and orientations. Consequently inferring an ancestral origin of such a protein is a difficult and challenging task. The need for clear evolutionary definitions is particularly acute for multidomain proteins, as their underlying coding sequences often have distinct, and even conflicting, evolutionary histories. Such proteins may share an inserted domain but are in fact unrelated. 45
A supplementary difficulty, known as chaining effect, may arise when constructing families of orthologs, as proteins including multiple domains may attract, via auxiliary domains, unrelated proteins (including different combination of domains).
Further difficulties are related to isofunctional genes and novel gene creation, which may lead to erroneously inferred orthology. Isofunctional genes are likely to share high sequence similarity, particularly at the protein level, and thus might be considered as homologs by sequence similarity methods, 46 leading to the erroneous detection of orthologs. The creation of novel genes is possible from noncoding sequences, as well as through domain shuffling, incorporation of mobile elements, or gene fission and fusion.47–49 Because homology is based on similarity criteria, new genes derived from noncoding sequences might be inferred as orthologs to true genes, whereas it is obvious that they are not: they do not share a common ancestor. To avoid this pitfall, such genes should be filtered out before applying any procedure for the search of orthologs.
Methods for Orthology Inference
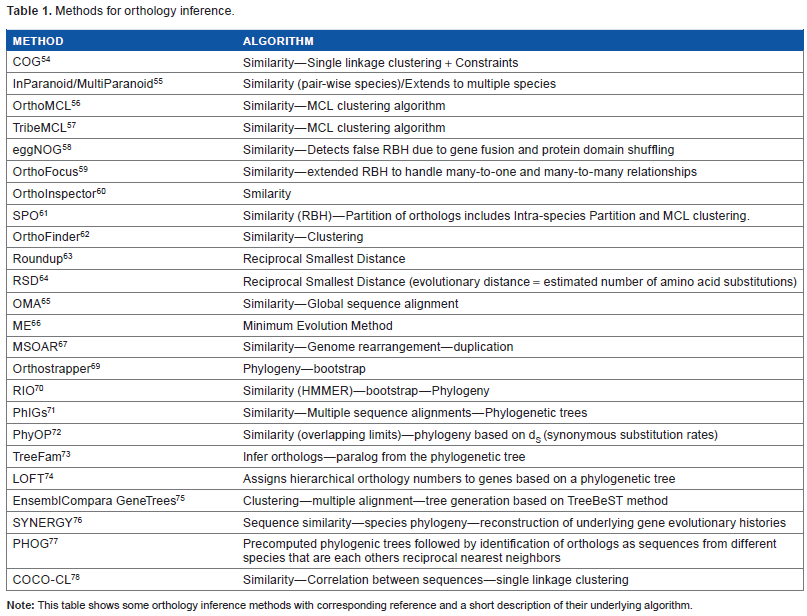
There are two main approaches to determine orthologous gene classes, based either on sequence similarity or on phylogeny (Table 1). 50 Comparative studies assessing the performances of different strategies by these methods have already been reported.17,19,51–53 Here, the salient features of some methods and their corresponding advantages and limitations are highlighted.
Methods for orthology inference.
Similarity approaches
Similarity-based approaches rely on genome comparisons and clustering of highly similar genes to identify orthologous groups. These approaches include the following: COG, 54 InParanoid, 55 OrthoMCL, 56 TribeMCL, 57 eggNOG, 58 OrthoFocus, 59 Ortholnspector, 60 SuperPartitions of Ortholog (SPO), 61 and OrthFinder. 62 The clustering of the set of inferred orthologs is generally based on classification criteria such as the single linkage method or the Markov Cluster Algorithm. 57 Other similarity methods based on evolutionary distance metrics criteria include roundup 63 (based on reciprocal smallest distance [RSD]), RSD, 64 OMA, 65 and a minimum evolution method. 66 A combination of sequence similarity, genome rearrangements, and duplication events is implemented in MSOAR 67 to identify pairs of orthologs in closely related species.
Similarity approaches relying mainly on reciprocal best hits (RBHs) are recognized to perform well in terms of comparative accuracy. Benchmark studies19,51,68 concluded that RBH methods outperform other approaches. However, the RBH approach was criticized for underappreciation of orthology in the presence of paralogy. More importantly, this simple approach was reported to suffer from conceptual drawbacks: (i) RBH analyses are restricted to the class of 1:1 orthologs, failing thus in the detection of many-to-(one and/or many) orthologs; (ii) the RBH approach may lead to overinclusiveness particularly when gene losses are involved in some of the considered genomes. 68 In such cases, a gene is erroneously considered as best hit because the real counterpart is lost; and (iii) the classification of orthologs detected with RBH methods is suspected to be prone to chaining effects in the motif and domain compositions. Such effects could be associated with gene fusion and domain shuffling events in multidomain proteins that evolved through different speciation and duplication events.59,61
Phylogeny approaches
Phylogeny-based approaches use candidate gene families determined by similarity and then rely on merging gene and species phylogeny to determine the subset of orthologs: Orthostrapper, 69 RIO, 70 PhIGs, 71 PhyOP, 72 TreeFam, 73 LOFT, 74 EnsemblCompara GeneTrees based on TreeBeST method, 75 SYNERGY, 76 and PHOG. 77 COCO-CL 78 is based on a hierarchical clustering algorithm of correlated genes guided by phylogenetic relationships. Species phylogeny was originally based on rRNA genes, and with the availability of complete genomes, it is now based on sets of shared genes. It should be noted that species phylogeny construction is still subject to debate, as discussed below.
Obtaining the correct phylogenetic gene tree and performing accurate reconciliation is crucial for the detection of orthologs. Phylogeny-based methods are deemed to be more reliable than similarity approaches, 79 but are difficult to automate because of the intrinsic weakness of the multiple alignment and phylogenetic tree construction methods that underlie gene phylogenies. Intrinsic weaknesses of multiple alignments include sequences of different lengths that require the introduction of gaps, and reshuffled sequences that lead to inaccurate alignments. 80 Moreover, the complexity of the phylogeny-based methods grows with the number of taxa, 81 particularly in large families where orthology is more difficult to assess. Such approaches are also subject to controversy on conceptual grounds, 82 as species phylogeny is not always straightforwardly established,9,16,61,83 with gene and species phylogenies not necessarily coinciding.
In practice, combination of the similarity and phylogeny-based approaches, together with manual annotation, helps to a rather reliable identification and clustering of orthologs. 84 This combination has been successfully illustrated by the reconstruction of gene histories in Ascomycota fungi. 85
Synteny conservation approaches
Although synteny describes the colocalization of loci on the same chromosome, synteny conservation is defined as the conservation of gene order and orientation along chromosomes 86 and constitutes a substantial source of evidence for determining gene ancestry. The adjacency of orthologous genes in different species provides reliable information to identify orthology relationships, because the comparison of closely related species revealed an extensive, quasi-integral conservation of gene arrangements along chromosomes.87,88 However, synteny conservation suffers from large interfamily evolutionary distances, asynchronous to the sequence divergence between orthologous genes, as shown for example in both drosophilids 89 and yeast species. 90
Similarity approaches were associated with conservation of synteny84,91,92 and neighborhood of genes between species93,94 to reliably detect orthologs in closely related species. The conservation of chromosomal environments has been used in specific methods to refine the identification of orthologous groups, as applied for example on the specific group of hemiascomycetous yeasts95,96 and vertebrates.
31
The information and conservation of gene arrangements help in improving the accuracy of orthology assignment and, in some cases, constitute the unique possibility to check for the reliability of detected orthologs. Figure 3 illustrates an example of a difficult scenario where a species S including a gene g that has been duplicated into g1 and g2, is followed by a speciation event, giving rise to two species S1 and S2, and by a gene duplication solely in S2 of g12 (resulting in g12a and g12b) and g22 (resulting in g22a and g22b). Two neighboring genes descendant of g0 and g3 are conserved throughout. In this situation, if genes g11, g22a, and g
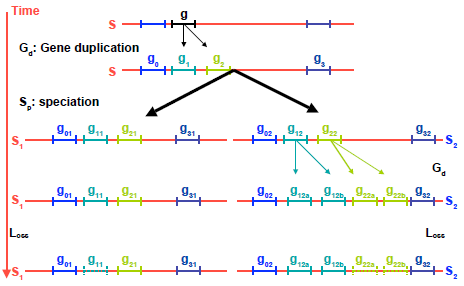
Example of a misleading situation in orthology inference.
Beyond sequence comparisons, synteny, and phylogeny-based methods, there are other, less traditional, methods that attempt to improve the prediction of orthologs and their functional analyses. 97 Some of these are based on large-scale analysis of protein–protein interactions and gene coexpression networks.98,99 A recent database, IsoBase, 100 resulting from function-oriented ortholog identification, seeks the integration of sequence data and protein–protein interaction networks to help identifying functionally related proteins.
Other methods based on shared protein domains have been suggested, but these methods are implicitly based on sequence similarity.101–103 Multidomain proteins pose a challenging question because of their ancestral origin particularly when they have undergone domain shuffling, and consequently on inferring their orthology. 45
SuperPartitions of orthologs
In large-scale proteome comparisons, a method called SuperPartitions 61 was introduced for ortholog inference and clustering into families called SPOs. The procedure is based on the partitioning of RBHs, with the further merging of partitions, including members of the same paralogous classes. This procedure is detailed in Ref. 61; here, only the main steps are summarized in the following section.
Distinct proteomes by pairwise comparisons lead to sets of RBH proteins that are considered orthologs. The set of all RBH proteins (deduced from the considered proteomes) is partitioned and each part is denoted as Pn.m (n being the number of distinct proteins in the part and m is an arbitrary index used to differentiate partitions containing the same number of proteins).
For each species, intraspecies comparisons lead to a set of nonunique proteins (proteins that have at least one hit in their own proteome) that are considered paralogs. These paralogous proteins are clustered according to their similarity using the mcl programme. 104 Each cluster is denoted as Cp.q (p being the number of proteins in the cluster and q is an arbitrary index used to differentiate clusters with identical number of elements). In each species, unique proteins, ie, with no hit in their own proteome are denoted as single.
Considering the whole set or orthologs obtained from all pairwise comparisons, each protein is part of an RBH partition denoted as Pn.m and of a cluster of paralogs in its own species denoted as Cp.q (or single if it is unique). Both classifications are joined to form the grouping category of each protein in the set of orthologs: Pn.m.Cp.q, ie, corresponding orthologous part and paralogous cluster.
RBHs partitions were further processed, by merging partitions including proteins belonging to the same intraspecies class Cp.q (labeled Pn.m.Cp.q), into a SuperPartition denoted as SPOr.s with r the number of proteins in the SPO and s an arbitrary order for indexing.
In order to facilitate the in-depth checking and study of predicted ortholog families (SPOs), the conservation profiles of the obtained clusters were used, which allow simple detection of SPOs containing duplicated members. For each SPO, the meme 104 suite of programs was used to search for shared motifs by all (or a subset of) protein sequences in the SPO, thus allowing the evolutionary structure of the members to be revealed (a practical example of this coding scheme is shown in Fig. 4).
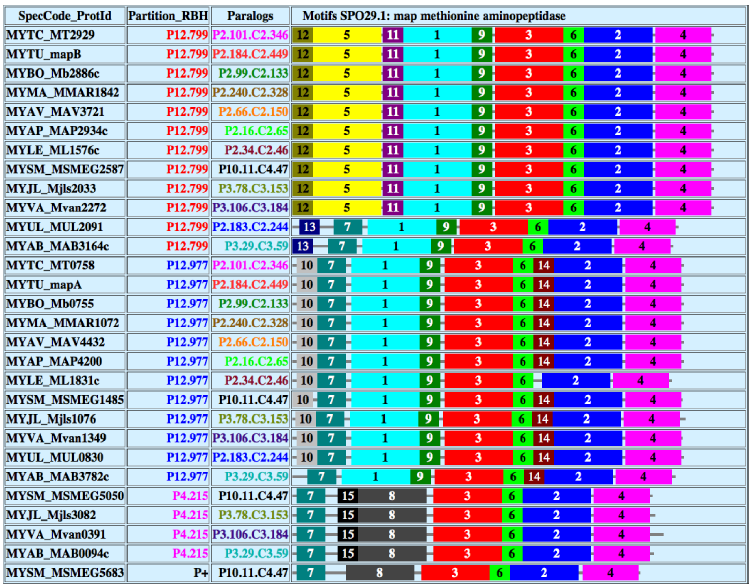
Assessment of members of orthologs in an SPO cluster by detecting motifs and their distribution.
As a result, members of a Superpartition (SPOr.s) were characterized (see Ref. 61 for the coding) by: (a) their corresponding RBH partition (Pn.m); (b) their intraspecies mcl cluster Cp.q (with the attached intraspecies partition Pn.m.Cp.q); and (c) their shared motifs as detected by meme. These detailed descriptions help in the checking and the assessment of orthology between the SPO members. In the example of Figure 4, four shared motifs appear in the same order within all members of the SPO, thus enhancing the validation of the predicted SPO members.
Perspectives and Open Questions
The search for orthologs is a milestone in the genome era, as its proper detection is crucial for evolutionary studies of genes and species. Comparative genomic analyses represent a powerful approach to recognize similarities between species, notably with the exponentially increasing amount of data generated by genome projects. Consequently, one of the primary tasks of evolutionary genomics is the determination of gene families from sets of taxa. The reconstruction of the evolutionary histories of genes and species relies critically on the accurate identification of orthologs. Such identifications are crucial for addressing a series of fundamental evolutionary questions concerning the determination of shared genes by different species, genes that share common core evolutionary history, or yet genes subjected to duplication, deletion, or transfer. The accumulation of these evolutionary events makes the reliable identification of orthologous genes a major challenge, particularly for distantly related species where traces of similarity are hardly recognizable.

Even though the manual detection of orthologs may be efficient for a small number of genes, automatic approaches are needed to deal with the large amount of genome data currently available. However, despite great efforts devoted to the development of orthology detection methods, the situation appears unsettled and to a large extent the quest for orthologs 17 is still an ongoing task in large-scale genome comparisons. The motivations of “The Quest for Orthologs” (http://questfororthologs.org/) consortium that is an open community are to benchmark, to improve accuracy and standardize orthology inference through collaboration, use of shared reference datasets (http://www.ebi.ac.uk/reference_proteomes), and evaluation of emerging new methods.
Species tree and ancestral sequence reconstructions may help to determine accurate orthologs. The species tree is crucial to delineate speciation events, whereas ancestral sequence reconstruction of actual sequences provides further information about their evolution over time. In the following section, some hints that may help methodological developments in inferring orthologs are pointed out.
Problematic Quality and Completeness of Whole-Genomic Data
Genome assembly and annotation
The starting point for orthologs detection is the availability of completely annotated genomes with their corresponding complete sets of genes; otherwise, it is unlikely to recover the correct full set of orthologs. For technical and methodological reasons, available genome sequences are of different quality: some are complete, while others are incomplete or in draft state (see statistics on: http://www.genomesonline.org/cgi-bin/GOLD/index.cgi). Genome data annotations are also of highly diverse quality. Apart from a few genomes, particularly from yeast,69,105 many of the available genome data are automatically annotated and have not been adequately checked. Moreover, annotations greatly depend on the quality of the sequences themselves. This is particularly serious for draft sequences, for which the lack of additional sequencing efforts to complete the assembly of chromosomes may mislead on the actual number of genes and the corresponding sequences, as quantified in Ref. 106 High-throughput sequencing technologies are now producing large amount of genomic data, raising fear of the low quality of assembly and annotation data. Consequently, an important emerging question is how accurate and useful are these data regarding orthology detection at a large scale?
This issue has been partially addressed in the case of model organisms. For example, a set of genomes have been extensively annotated, with the aim to study genes showing specific functions involved in the adaptation of species in specific environments (see Ref. 107 for the Archaea example) and to be used as reference for homology annotation of genes from closely related species. Only a few distantly related species received particular attention concerning this matter, from the human and teleostan fish genomes to the bacteria Escherichia coli and Bacillus subtilis, including the yeast Saccharomyces cerevisiae and the ciliate Paramecium tetraurelia. These efforts confidently allowed the automatic annotation of a number of closely related species, but rapidly have shown their limits when large-scale comparisons have been considered. As an illustration, the rate of erroneous annotations has been measured to be >30% in the UniProt/Swiss-Prot database. 108
Taxon sampling and species coverage
With the exception of the model organisms mentioned earlier, the accurate selection of sequenced genomes is still a pending question. While a number of scientific communities focus on very specific groups of monophyletic species as for example the 1000109 and 1001110 genome projects for Homo sapiens and the plant A rabidopsis thaliana, respectively, the great majority was attached to sequence and characterize distantly related species, from known or unknown taxonomic groups, with the aim of extending our knowledge on the tree of life. This is the case for example in yeast for the Dikaryome project that aims to extend the known phylogeny to the Dykarya phylum. 111 This bias is important in orthology identification using phylogenetic methods, and obviously penalizes automatic discrimination between true orthologs and horizontally acquired genes. 112 False bacterial hits are favored in the case of eukaryotic genes, and erroneous most similar hits in the case of bacterial genes. Indeed, the biased distribution of proteins toward bacteria and model organisms in current databases artificially increases the chance to find bacterial (rather than eukaryotic) genes as the most similar sequences to the one under study.
The reconstruction of syntenic blocks, chromosomal segments in which the order and orientation of the genes are conserved through the evolution of corresponding species, is also limited by the correct taxon sampling of the species under study. In closely related species, a large part of genes is still found in short syntenic blocks as for example in fly and in yeast.89,90 However, at large evolutionary distances, it remains very difficult to accurately define conserved segments, due to frequent events of synteny breakage. Consequently, one has to choose an appropriate set of closely related species for the analysis of synteny conservation, and hence for the reconstruction of ancestral genomes.
Therefore, the taxon sampling issue has a decisive impact on both phylogenetic tree and synteny reconstruction, thus affecting the identification of orthologous sets of genes. This could be partly solved by the correct choice of sequenced species, in this way filling the gap of uncovered phylogenetic regions and decreasing the span between compared species.
Ancestral sequence reconstructions and species tree
Reconstructions of ancestral gene sequences and evolutionary events traditionally reported by species trees are key to orthology detection and validation. In silico reconstruction of ancestral gene, protein, and chromosome sequences provides information about evolutionary events. For example, conserved genomic islands identify features that are protected from variation by natural selection. When an ancestral sequence is predicted for a set of actual sequences (genes, chromosomes, or genomes), multiple alignments of the predicted ancestor and the actual sequences should provide hints about mutations (substitutions), deletions, and insertions that took place during the evolutionary time in these sequences. Such reconstructions might give indications about gene gain or loss, as well as genome reduction. Consequently, ancestral prediction represents one of the expected results that may validate orthology detection as shown by ongoing efforts in algorithmic development6,85,113–116 and particularly when such reconstructions are performed in association with species tree topologies, shedding light on specific evolutionary events at a given node of the tree. Optimal strategies for ancestral sequence and genome reconstructions are still under development, evaluation, and debate.30,117–120
Building the tree of life genome by genome,13,14,121 and tracing the tree of life, 122 together with advances in methodological tree of life construction 9 will allow the setup of an accurate species tree topology. Because of the observed incongruence between gene tree and species tree topologies, specific methodological developments to reconcile these topologies have been initiated to delineate the degree of confidence in such tree topologies.123–128 The reconciliation processes usually involve hypotheses on gene duplications and losses in order to account for the topological incongruences.
The assessment of such theoretical models in constructing species trees can benefit from the availability of recently discovered data and new resources. In this regard, the availability of ancient DNA sequences might be a major resource in filling gaps between distantly related species on the species tree and allows for better estimation of speciation events. 129 Other significant resources are shown by an interesting example, 130 which traced the first step to speciation in a salamander population.
Experimental validation of predicted sets of orthologs?
While the vast majority of published orthology sets only relies on computational analysis of gene sequences without further experimental validation, functional similarities can be assessed assuming the absence of paralogs in the considered groups of genes. 131 One approach is the heterologous replacement of a particular gene by its ortholog from another species. This experimental setup allows measuring the complementation of the function of a gene by its orthologous gene, by complementing mutant cells or restoring a particular phenotype. Only a few cases of orthologous relationships have been validated using this type of isofunctionality testing in S. cerevisiae mutants.132,133
Isofunctionality can also be assessed by analyzing the similarity of interacting partners within protein interaction networks. In vitro two-hybrid experiments could reveal the biological proof for the existence of orthologous genes. In this case, pairs of purified gene products are tested for interaction, measured by two-hybrid essays. This method has long been used to establish interaction maps within species 134 and could be adapted to cross-species comparisons. Once the interaction between two proteins is verified within one particular species, the positive match after replacement by an orthologous protein from another species could provide direct evidence of functional complementation, whereas the negative match does not necessarily imply absence of orthology.
With the exception of few cases, the proofs for reliability and completeness of predicted groups of orthologs in a set of species are still pending questions. Indeed, a list of orthologs in a set of species is trusted as long as the corresponding species phylogeny is trusted.
At the structure level, relationship between sequence similarity and structural similarity has long been established, but little is known about the impact of orthology on the relationship between protein sequence and structure. 135 It has been shown that orthologous proteins exhibit a greater similarity of domain architectures (ie, domains structure along a sequence) as compared with paralogous proteins at the same level of similarity.135–137 This result is confirmed by the comparison of orthologs and paralogs with the available crystal structures. 135
In this regard, it is interesting to note that the conservation of sequence, structure, or genomic context is not implicit in the definition of orthology. 137
A significant issue on reliability is the distribution of predicted orthologs (or conservation profile) 61 in a given cluster, conveying important information about the content, expansion, and reduction of such a cluster among the surveyed species. In this regard, only a few reports have established a possible gain or loss of members in a given cluster of orthologs by considering systematic studies in specific situations.6,85 The work of Trachana et al 53 focused on the assessment of accuracy of the methods in ortholog assignments by considering 70 manually annotated protein families, and hence the possibility of comparing the ability of each method to detect orthologs in a given family.
For practical reasons, there is a need for a public standard set of genomes that can be used to compare methods in predicting orthology. This set should include the typical difficulties discussed earlier and could be used to test the sensitivity and selectivity of orthology detection methods. This concept has already been discussed,138,139 and sets of reference proteomes (http://www.ebi.ac.uk/reference_proteomes/) and orthologycurated databases (http://questfororthologs.org/orthology_databases and http://eggnog.embl.de/orthobench2) to be considered for ortholog assignments and evaluation have been suggested. Unfortunately, while these sets are useful to show the differences between orthology detection methods, they are insufficient to estimate their respective accuracy with regard to the aforementioned difficulties (including HGT, duplication, loss, and proteins with multidomain ancestry, among others).
An optimal standard set should include both closely and more distantly related species sampled from the species tree. The selected species should correspond to the pointed difficulties in detecting orthologs, including gene loss, HGT, duplication, WGD (one or several rounds), and genome reduction. Analyses of this set by orthology prediction methods should illustrate their corresponding ability to detect a given difficulty and their accuracy in identifying the correct set of orthologs and their clustering.
Why do so many orthology predicting methods exist?
At this point, one may wonder why so many methods exist (although only a few of them are cited here). The simple answer is that there is still no evidence on how to deal with the complex evolutionary events that have been mentioned in this work and that hamper correct orthology detection. Current methods have been developed to overcome particular difficulties, but none appears to be reporting universal solutions.
Cross-comparison studies of orthology detection methods17,19,51–53,68,97 show significant differences in their corresponding sets of orthologs, and even contradictory results between large-scale studies that evaluate the relative algorithms. 68 It has been reported 97 that popular methods show <50% of concordance in establishing overlapping sets of orthologs, and even <30% when distantly related species are taken into account. These authors further suggested that a deeper understanding of the error-prone steps in the algorithms could trigger developments toward better ortholog detection and clustering, by focusing on inconsistent sets of orthologs predicted by different methods. They conclude that “challenges for RBH-based approaches center around how to reduce false positives. In contrast, phylogeny-based approaches have many more aspects to consider including: selection of genes to build the tree and the accuracy of the tree reconciliation with known phylogeny.”
In practice, the combination of methods (similarity and phylogeny based) together with the known organization of the considered genomes (particularly concerning synteny conservation, introgression, and segmental and whole-genome duplication) are currently the best procedure to enhance the validity of the inferred set of orthologs and paralogs.
Concluding Notes
This review has reported some of the many available methods in inferring orthologs and their clustering. The evolutionary dynamics involving duplication, loss, fusion/fission of genes, and segmental and whole-genome duplication are some of the difficulties that hamper the detection and clustering of orthologs in a large set, including distantly related species. The review has also mentioned the contribution of methodological developments and resources that may help establishing a species tree that is at the core of reliable orthology detection.
Appropriate sequencing and annotation efforts in sets of sampled species should provide reliable sets of orthologs when using a combination of similarity and phylogeny methods together with prior knowledge related to evolutionary dynamic events. Large-scale orthology detection from distantly related species can be approached in two steps, first on locally (at the taxon level) reliably defined clusters of orthologs that can then be assembled in a second step to cover the whole large set of species.
Finally, it is suggested to filter out, prior to performing large-scale orthology inference, the considered proteomes from proteins resulting from HGT and to fix possible difficulties related to domain shuffling, gain, and loss.
Key Points
This review describes some available methods in detecting orthologs between species, with brief indications about their advantages and limitations.
Orthology detection is hindered by the evolutionary dynamics, including duplication, transfer and loss of genes, and shuffled multidomain proteins, as well as by the questionable quality of available genome data in terms of completeness and annotation.
It is suggested that orthology detection at large scale should be first performed locally at the taxon level, where existing methods and manual validation generally result in reliable detection and clustering of orthologs, and then assembled following the species tree as a template.
Methodological developments are still needed in the automatic inference and clustering of orthologs in conjunction with species tree construction, as both concepts are tightly linked.
Author Contributions
Conceived the concepts: FT. Analyzed the data: FT. Wrote the first draft of the manuscript: FT. Developed the structure and arguments for the paper: FT. Made critical revisions: FT. The author reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
I thank Bernard Dujon for constant support, Pedro Alzari for his careful reading of the manuscript and his suggestions, and Thomas Rolland for constructive discussions.