
Abstract
MethyLight is a sodium-bisulfite-dependent, quantitative, fluorescence-based, real-time PCR strategy that is used to detect and quantify DNA methylation in genomic DNA. High-throughput MethyLight allows the rapid and sensitive detection of very low frequencies of hypermethylated alleles in populations of alternated individuals. The high sensitivity and specificity of MethyLight can be applied not only to make it uniquely suited disease clinical but also quantitatively assessed of these low-frequency methylation events. Owing to its full of advantages of simple procedure, high efficiency and high sensitivity, MethyLight provides a powerful approach for clinical examination, Gene expression analysis, SNP analysis and allele analysis. Coupled with other techniques, MethyLight can be used immediately in identifying allelic alterations in genes exhibiting expressions correlating with phenotypes, Locating an allelic series of induced point mutations in genes of interest. The development of this technique should considerably enhance our ability to rapidly and accurately generate epigenetic profiles of samples.
Introduction
Epigenetics can be defined as the study of changes in the regulation of gene activity and expression that are not driven by gene sequence information. Epigenetic alterations are now well recognized as highly relevant to many common diseases.1,2 The area of DNA methylation has grown dramatically and has become one of the most dynamic and rapidly developing branches of molecular biology for Epigenetic analysis over the last decade. This has resulted in a very urgent demand to develop a high throughput technique that is a simple procedure, highly efficient and highly sensitive, and capable of finding a wide range of mutant alleles that is needed for further study of functions analysis. Conventional genetics can hardly meet the demands of high throughput and large-scale surveys of gene function. The assay as we have explored it here is highly quantitative, highly efficient and highly sensitive. Rather than capturing all methylation occurrences of a CpG dinucleotide in a heterogeneous genomic DNA sample, the MethyLight technique can accurately determine the relative prevalence of a particular pattern of DNA methylation. However, in doing so, the technique is oblivious to all other methylation permutations. MethyLight determines the relative amounts of a particular methylation pattern with quantitative accuracy. Whereas the quantitative nature of MethyLight is based on the cycle number at which the fluorescent signal crosses a threshold in the exponential phase of the PCR reaction. This allows quantitative conclusions to be drawn concerning methylation levels relative to a control reaction. The most striking advantage of MethyLight, as compared to existing techniques, is its potential to allow the rapid screening of hundreds to thousands of samples. Unlike other techniques, the MethyLight assay is completed at the PCR step, without the need for further gel electrophoresis separation or hybridization. This reduces the chance of sample and dramatically decreases the amount of labor involved in DNA methylation analysis. High throughput MethyLight allows the rapid and effective detection of induced point mutations in populations of mutation individuals, and help locate an allelic series of induced point mutations in genes of interest. This makes MethyLight an attractive strategy for a wide range of applications for basic functional Genomic study.
Development of MethyLight
The technology of MethyLight was first reported by Eads, C. A and colleagues at Department of Surgery, University of Southern California School of Medicine, Norris Comprehensive Cancer Center in Los Angeles, CA, in the 2000s. The precision and performance characteristics of MethyLight was demonstrated by quantitative DNA methylation analysis in a study in which the authors used MethyLight to measure percentage of methylated reference (PMR, i.e. degree of methylation) for the MGMT, MLH1, and CDKN2A (p16) promoters to assess run-to-run variation from the pooled DNA extracted from each individual person. The PMR method is used as a general measure of DNA methylation, since it controls many other sample-independent sources of experimental variations and errors. The formula to calculate PMR values represents the quotient of two ratios (x100). Thus, the formula is: 100 x [(GENE-X mean value) sample/(ALU mean value) sample]/[(GENE-Xmean value) M.SssI/(ALU mean value) M.SssI]. Once the real-time PCR program is finished, the Ct values are converted to mean values/copy numbers using the standard curve for each plate. One PMR value per sample will be calculated based on the mean values derived from each of the two standard curves. The two PMRs obtained will be averaged at the end of the procedure. Using the data generated with the first standard curve, divide the mean/copy value for the methylation reaction of the sample of interest by the mean/copy value of the ALU reaction. (Methylight Chapter 23) Because MethyLight technology is a scaled up validation, highly efficient and highly sensitive of Epigenetic strategy, it has been rapidly developed. Initially MSP used to detect aberrant methylation patterns in human samples with substantial contamination of normal DNA, such as non-microdissected, heterogeneous tissue samples. A Sensitivity and quantitative accuracy of MethyLight technology was published later, which used DNA oligonucleotides that anneal differentially to bisulfate-converted DNA according to the methylation status in the original genomic DNA by the quantitative, fluorescence-based, real-time PCR suited for this application.3,4 In 2001, the standard proposal was developed, and the PMR calculations were explored, so the MethyLight technology has become the routine method to detect mutations and satisfactory results have been obtained. 4
In the first step of the high throughout MethyLight process, purified genomic DNA was treated with sodium bisulfite according to established protocols and analyzed using a real-time MSP assay, briefly, 50 uL of DNA was denatured by adding 5.5 uL of 2 M NaOH for 10 min at 37° C. Next, 30 uL of 10 mM hydroquinone (Sigma) and 520 uL of 3 M sodium bisulfite (Sigma) at pH 5, both prepared fresh, were added. Samples were then layered with mineral oil and incubated at 50° C overnight. Modified DNA was then purified using the DNA Wizard Clean-Up Kit (Promega) according to the manufacturer's protocol and eluted with 50 uL of water. Chemical modification was completed by treating DNA with 5.5 uL of 3 M NaOH and incubating it for 5 min at room temperature. DNA was precipitated with etha-nol and resuspended in 20 uL of water and stored at 20° C until used.
5
This generates methylation dependent sequence differences at CpG dinucleotides by converting unmethylated cytosine residues to uracil, while methylated cytosine residues are retained as cytosine. After sodium bisulfate conversion, genomic DNA is amplified by fluorescence-based, real-time quantitative PCR. In brief, bisulfite-converted genomic DNA is amplified using with a 5′ fluorescent reporter dye (6FAM) and a 3′ quencher Dye (TAMRA).6,7 the 5′ to 3′ nuclease activity of
Advantages of the Methylight assay
Simple procedure
Molecular genetics have shown that DNA methylation is associated with gene silencing and plays an important role in the developmental process such as X-chromosome activation and genomic imprinting. Subsequently, considerable advances have been made in high-throughout technology for DNA Methylation. The first generation of methylation detection assays employed the digestion of genomic DNA with a methylation-sensitive restriction enzyme followed by either Southern blotting analysis or PCR. 8 However the limited availability of informative restriction sites, the occurrence of false positive results due to incomplete digestion, and the requirement of large amounts of high molecular weight DNA have restricted their use. A second generation of techniques resulted from the demonstration that treatment of genomic DNA with sodium bisulfate followed by alkaline treatment converts unmethylated cytosine to uracil. 9 While leaving methylated cytosine residues intact, sequence variants at a particular locus can subsequently be analyzed by PCR amplification with primers designed to anneal with bisulfite-converted DNA. The sequence differences resulting from various DNA methylation patterns can then be revealed in two principally different ways (Fig. 1). The MethyLight is a combination of two technological advances-bisulphate modification of DNA and methylation-specific polymerase chain reaction (MSP). 10 The benefit of sodium bisulfite-based assays is that they require very small amounts of DNA, and consequently are compatible with DNA obtained from samples that have the amount of availability of informative restriction sites.9,11,12 Methylation-specific polymerase chain reaction (MSP) avoids using electrophoresis and employing restriction enzyme digestion, radio labeled dNTPs or hybridization probes. This technique requires no complicated post-manipulation and expensive devotion. It can also allow for rapid analysis of many samples at multiple gene loci and can be systematically investigated for gene function.
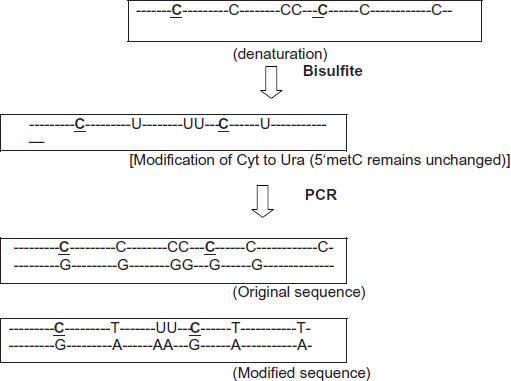
Sodium bisulfite conversion. Sodium bisulfite modifies the sequences of genomic DNA by converting unchanged Cyt to Ura while leaving 5metC unchanged. PCR amplification results in the replacement of Ura by Thr. So two different PCR products can be generated: the original sequences and the modified sequences).
High sensitivity
The high mutation-detecting efficiency of MethyLight was given in the original work by Cindy A Eads and his colleagues.
3
In this thesis, ESR1 (the interest gene) methylation can be detected reliably in the presence of a 10000-fold excess of unmethylated alleles by MethyLight technology. MSP is sensitive to 0.1% methylated alleles of a given CpG island locus, and can be performed on DNA extracted from paraffin-embedded samples.
10
Human sperm DNA that had been fully methylated by treatment with SssI methyltransferase
High efficiency
The high methylation–detecting efficiency of MethyLight is attributed to its high frequencies of ‘CG’ islands throughout the genome and the densities of ‘CG’ islands mutation could be estimated. For example, Subjection of denatured DNA to sodium bisulfite results in the conversion of cytosine residues to uracil, while methylated cytosine remains unaffected. This is assuming that all changes are C/G to T/A transitions. Based on these results, the most suitable methylation site is selected in a specific gene of interest. This includes the ability of Sodium bisulfite conversion to induce high density methylation in multiple loci so that genome-wide saturated methylation can be achieved using a relatively small mutant population. The frequencies of methylation is different in various species and receptors. On the basis of the above estimation, a total of 10000 methylation sites will achieve satisfactory methylation densities.
MethyLight does not require post-PCR manipulations of DNA such as gel electrophoresis, mainly because the analysis is performed at the PCR level. All current methods of bisulfite-based DNA methylation analysis rely on subsequent PCR amplification. The real-time PCR technology provides MethyLight a very powerful application capacity. For instance, the automated manipulation has been realized in the MethyLight, 13 and the methylation status of six different CpG dinucleotides is interrogated with six CpG dinucleotides, there are 26 = 64 different permutations of methylation status. If both of the primers and the probe each overlap two CpGs, then the total number of variants contained within the sequence covered by the oligonucleotides is 4 x 4 x 4 = 64. In theory, one could design separate PCR reactions to analyze the relative amounts of each of these potential 64 sequence variants. And also MethyLight can be used to direct quantization of methylation at the PCR amplification step in closed-tube reactions, thereby reducing the potential for contamination and eliminating further manipulations such as gel electrophoresis and autoradiography. Moreover, the MethyLight product length is typically designed to be fairly short (50–200 bp) because PCR amplification-Primer and Probe Designation and consequent fluorescence emission become inefficient with the longer extension time that is required (Fig. 2). The result is a substantial improvement in throughput capability owing to the optimums which are mentioned above. So automation of MethyLight assays is possible, and numerous samples or loci can be rapidly and simultaneously analyzed.

Methylation-specific PCR (
Application of MethyLight
For Clinical diagnosis
The MethyLight technique was first utilized in cancer detection. P16 and p15 gene methylation in head and neck squalous cell carcinoma and their quantitative evaluation in plasma were studied. 14 The differential levels of methylated p16 and p15 DNA in plasma might be potentially useful markers in screening high-risk populations for early HNSCC and monitoring their treatment response. Through the workshop, mutant materials, DNA samples and mutant information were fully shared by all researchers working on epigenetic markers. It is said that Tampons can be used as a new tool to detect endometrial cancer and collect Methylated DNA. 15 The methods developed in this study provide the basis for a prospective clinical trial to screen asymptomatic women who are at high risk for endometrial cancer. A new prostate cancer marker can be developed subsequently, which might increase the accuracy of early detection, diagnosis, and prognosis prediction, constituting an attractive and fast-growing research fields, 16 in which CDH1 and CDH13 methylation in serum is an independent prognostic marker in cervical cancer patients. 17
Epigenetic markers have proved to be a successful case for the application of MethyLight in cancer detection and have encouraged the broader utilization of the technique to other organisms. Well-developed and tested protocols have been available for cancer detection, such as Alu Repetitive elements, 18 and LINE Repetitive elements.19,20 LINE-1 elements are usually methylated in somatic tissues, and LINE-1 hypomethylation is a common characteristic of human cancers.21–23 Hypermethylation of CpG islands occurs in a non-random fashion in cancer cells, and the DNA methylation patterns observed appear to be tumor specific, suggesting that gene-specific methylation events represent potentially useful markers for molecular diagnostic testing in cancer. The transcriptional silencing of tumor suppressor genes by promoter CpG island hypermethylation can contribute to oncogenesis. 20 CpG island methylation, responses to combination chemotherapy, and patient survival in advanced microsatellite stable colorectal carcinoma was firstly used with MethyLight technology in 2007 by Ogino, S and colleagues. 24
The MethyLight assay was used to quantitate the methylation of CpG islands within the MLH, P16 (INK4 A), TIMP3, DAPK, APC, ER and MYOD genes. A real-time, methylation-specific polymerase chain reaction assay was also used to quantitate the methylation of LINE-1 repeats. 20 Moreover, Alu sequences are also normally methylated in somatic tissues,25–27 and are thought to become hypomethylated in human cancer cells. Several studies have reported associations between DNA methylation markers and response to chemotherapy.28–30 The CpG island methylation phenotypes (CIMP or CIMP-high) with extensive promoter methylation is a distinct phenotype in colorectal cancer. However, a choice of markers for CIMP has been controversial. So Model and his colleagues have detected identification and validation of colorectal neoplasia-specific methylation markers for accurate classification of disease. A recent extensive investigation has selected five methylation markers (CACNA1G, IGF2, NEUROG1, RUNX3, and SOCS1) as surrogate markers for epigenomic aberrations in tumors. 31 In this series, a panel of markers including at least RUNX3, CACNA1G, IGF2, and MLH1 can serve as a sensitive and specific marker panel for CIMP-high. In conclusion, such prospective clinical trials using DNA methylation markers have yet to be conducted, nevertheless, a flood of reports on predictive DNA methylation markers is predicted in the near future.
For DNA Polymorphism assessment
DNA polymorphism far and wide exists in a variety of species and plays an important role in biological evolution. Many methods currently are available for revealingDNApolymorphismsuchasDNAsequencing, single-strand conformation polymorphism (SSCP), hybridization, and microarray, and these methods have their own advantages and disadvantages. Although DNA sequencing is simple and straight-forward, it is rather costly and time-consuming. SSCP provides a high-throughput strategy for polymorphism detection; however, it has low efficiency in detecting novel mutations with a limit of 200 to 300 bp length of target DNA sequence. Microarray holds two disadvantages, one is high cost of operation, and the other is the low detecting-frequency of less than 50%. Based on MethyLight, a strategy, referred to microarray derived MeDIP-enrichment was developed to detect DNA polymorphism present in naturally occurring mutations.32,33 MEDME 33 (modeling experimental data with MeDIP enrichment) can detect DNA variations from single nucleotide polymorphism(SNP), MEDME and can be performed as a high-throughput, low-cost, and high-accuracy approach compared with the other methods mentioned above. It can simplify estimate the interpretation of the results both at single-loci and at chromosome-wide levels. It is very cost-effective and requires only a small proportion of the whole cost of the conventional approach of sequencing a genetic locus in every individual. By using MEDME Mattia Pelizzola and colleagues proposed that it can evaluate the true relationship in a high-throughput setting and a model-based analysis to predict the absolute and relative DNA methylation levels. In this experiment they have evaluated DNA methylation status of normal human melanocytes compared to a melanoma cell strain.
For Gene expression
MethyLight is relied on the TaqMan system
34
and the application of fluorescent probes,
35
So it appears commonly convenient, rapid and more accurate for mRNA detection used than the previous methods such as cDNA chips and differential display, The shortcomings of both technologies can only be qualitative rather than a quantitative analysis of the product, And with the development of biotechnology, a number of the corresponding kits are introduced, Which will ensure the high-throughput MethyLight in aspects of applications. The emergence of quantitative PCR technology will undoubtedly provide a great convenience for detection of the product and give a more complete and accurate result than cDNA chips and cDNA differential display technology. Gene expression is a dynamic process and is tightly connected to changes in chromatin structure and nuclear organization.36,37 So our ability to understand the intimate interactions between proteins and the rapidly changing chromatin environment will require methods that will be able to provide accurate, sensitive, and unbiased mapping of these interactions
Perspective
As a highly specific, sensitive reproducible and high throughput technique, MethyLight has been put into practice because it can rapidly detect biologically relevant information in the samples. It has been convincingly proved that MethyLight requires only minute amounts of DNA of modest quality, making it compatible with small biopsies and paraffin-embedded tissues. It represents an extension of the use of methylated and unmethylated permutations in samples and rapidly generates biologically relevant information with a minimal amount of manual labor and allows direct identification of beneficial genes and SNP single site changes in genes with known functions. The range of alleles that can be developed by methyl-chip in a short time is unparalleled and unlikely to be found elsewhere in the pool of genes, so the results of basic scientific research can be efficiently translated into crop improvement as new information about the functions of potential gene targets becomes available.
There are at least two immediate applications in Cancer detection using MethyLight and MEDME as a haplotyping tool for detection of genetic loci that are putatively associated with clinical important traits. The first application is the identification of allelic variation in genes exhibiting expression correlating with phenotypes. This will link gene expression with DNA variation. Because haplotypic variation caused by SNP or small indels can be detected, it can help overcome the main difficulty of finding DNA variation based on restriction-site polymorphism or linkag to hypervariable markers such as SSR. The second application is the establishment of an allelic series at genetic loci for the traits of interest in germplasm or induced mutants. Allelic series at such loci will provide confirmatory evidence of the relationship between the phenotypes and candidate gene sequences. A large collection of alleles at a locus will provide patterns of association to infer the functional significance of certain SNPs.
It should be emphasized that the MethyLight technique was not designed to yield high-resolution methylation information, such as the pattern information obtainable with bisulfate genomic sequencing or the accurate methylation percentage determination at single CpGs obtainable with convention assay. As such, it should extend and complement ongoing efforts to determine molecular profiles of samples through high-throughput genomic and RNA-based technologies. It has been suggested that the recent progress in the area of plant molecular biology and plant genom-ics have the potential to initiate a new technology revolution. However, this technology needs to be implemented in new cultivars so it can realize this potential. MethyLight, as a unique technology for epigenetic profiles, coupled with other recently developed genomic resources, can be predicted to have more and more direct or indirect benefits that will be revealed through continuous applications of MethyLight in the near future. For example “epigenetic drugs”41–44 will be come into existence and demethylating agents will be used to treat patients with myelodysplastic syndrome (MDS). In addition MethyLight technology will contrive looking at genome-wide studies of histone modifications and of DNA methylation as well as chromatin remodeling in its entirety. Integrated epigenetic maps will be developed with a huge analytic capability being developed. It can also be predicted that more and more direct or indirect benefits will be revealed through continuous applications of MethyLight in the near future.
Abbreviations
PCR, Polymerase chain reaction; PMR, Percentage of methylated reference; MDS, myelodysplastic syndrome; SNP, Single Nucleotide Polymorphism; MEDME, Modeling experimental data with MeDIP enrichment; CIMP, The CpG island methylator phenotype; HNSCC, Head and Neck Squamous Cell Carcinomas; DamID, DNA adenine methyltransferase identification; MGMT, O6-methylguanine-DNA Methyltransferase; MLH1, Human mutL homolog 1; CDKN2A, Cyclin-depen-dent kinase inhibitor 2A, MSP, Methylation-specific polymerase chain reaction; SSCP, Single-strand conformation polymorphism; ESR1, Estrogen Receptor 1; CDH1, Cadherin 1; CDH3, Cadherin 3; TIMP3, Tissue inhibitor of metalloproteinase 3; DAPK, Death-associated protein kinase; APC, Ade-nomatosis polyposis coli; ER, Endoplasmic Reticu-lum; MYOD, Myogenic Differentiation Antigen; CACNA1G, Alpha1G T-type calcium channel gene; IGF2, Insulin-like growth factor 2; NEUROG1, Neurogenic differentiation 1; RUNX3, Runt-related transcription factor 3; SOCS1, Suppressor of cytokine signaling 1.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Acknowledgements
This research was supported by the Program for the Science and Technology department application foundation of Sichuan Province (2006J13-039), and The Changjiang Scholars and Innovative Research Team at the University of China (Grant No. IRT-0453) and the Program for Ministry of Agricultural of the People's Republic of China (2003-Q03) and the Program for the Ministry of Science and Theology of the People's Republic of China (2006BAD13B03) and High-tech Research and Development Program of China (2006AA100103).