
Abstract
The terms bioaccumulation and bioconcentration refer to the uptake and build-up of chemicals that can occur in living organisms. Experimental measurement of bioconcentration is time-consuming and expensive, and is not feasible for a large number of chemicals of potential regulatory concern. A highly effective tool depending on a quantitative structure-property relationship (QSPR) can be utilized to describe the tendency of chemical concentration organisms represented by, the important ecotoxicological parameter, the logarithm of Bio Concentration Factor (log BCF) with molecular descriptors for a large set of non-ionic organic compounds. QSPR models were developed using multiple linear regression, partial least squares and neural networks analyses. Linear and non-linear QSPR models to predict log BCF of the compounds developed for the relevant descriptors. The results obtained offer good regression models having good prediction ability. The descriptors used in these models depend on the volume, connectivity, molar refractivity, surface tension and the presence of atoms accepting H-bonds.
Keywords
Introduction
Bioaccumulation is the process where the chemical concentration in an aquatic organism achieves a level exceeding that in the water, as a consequence of chemical uptake through all means of chemical exposure (e.g. dietary absorption, transport across the respiratory surface, dermal absorption, and inhalation). Bioconcentration is defined as the absorption of a chemical concentrations in an aquatic organism's tissues that is greater than that in the water as a result of exposure of the organism to a chemical concentration in the water via non-dietary routes. The extent to which a contaminant will concentrate in an organism is articulated as BCF which is the ratio of the chemical concentration in the organism (CB) and the water (Cw)1:

Within a species, BCFs vary for different chemical compounds. The BCF of a chemical is the result of four processes: Absorption, Distribution, Metabolism and Excretion (ADME). 2 Bioconcentration is usually derived under laboratory conditions where the chemical is absorbed from the water via the respiratory surface and/or the skin only. Generally, the experimental measurement of bioconcentration is time-consuming and expensive, and is not feasible for a large number of chemicals of potential regulatory concern. For this reason attention is turning to estimation of BCF values by QSPRs. BCF values can be estimated from the octanol/water partition coefficient (Kow) using QSPR models. In addition, Kow values, either experimentally determined or estimated, can be used directly to assess the potential for bioaccumulation.
QSPRs are usually observed between log BCF and log Kow. The majority of these are linear regression models3–8 which give satisfactory results only for chemicals with log Kow < 6, while for highly hydrophobic chemicals (log Kow > 6), non-linear,9,10 bilinear11,12 or polynomial 13 relationships have been proposed and applied for more satisfactory BCF prediction/estimation. Zhao et al 14 performed a QSPR study on a large set of 473 compounds that was created with ISIS BASE 2.5 SP2. Lu 15 proposed models for the prediction of BCF using connectivity indices and comprising experimental BCF data in fish (at steady-state) for 239 non-ionic organic compounds. This approach was applied by Gramatica and Papa16,17 with the same data set previously modeled by Lu 15 using a large starting set of theoretical molecular descriptors and by applying a genetic algorithm (GA) approach as the variable subset selection method to obtain multilinear regression models with few variables correlated with BCF. Fatemi et al 18 reported a QSAR study on a set of 53 compounds using geometric, electronic and topological descriptors by artificial neural networks (ANN) to predict the value of log BCF for some compounds. Recently, a QSAR model for fish BCFs of 8 groups of compounds was developed employing partial least squares (PLS) regression, based on linear solvation energy relationship (LSER) theory and theoretical molecular structural descriptors.19–21
Khadikar et al 22 reported QSAR study on the estimation of bioconcentraction factor of polyhaloge-nated biphenyls using a distanced-based index called Padmakar-Ivan index, abbreviated as PI. Incited by these results, we have used the molecular descriptors used by Lu et al 15 Gramatica and Papa16,17 and Khadikar et al 22 in addition to large set of other topological indices to model the BCF. It is worth to mention that, to our knowledge and to date, no attempt has been made to investigate neural network modeling using principal components for prediction of BCFs of the set of nonionic organic compounds.
Before discussing our results it is necessary to mention that both Lu et al
15
and Gramatica and Papa in16,17 used the same data set of 238 compounds, while Khadikar et al
22
used only 16 polyhalogenated biphenyls from the original set of 238 compounds reported by Lu et al.
15
Furthermore, Gramatica and Papa16,17 did not verify the data and only deleted acrolein from the original data set reported by Lu et al.
15
In this contribution, we observed that 9 compounds (compounds:
In view of the above, we used principal components–-artificial neural networks (PC-ANN) and PLS methods on the data set of 229 non-ionic compounds modeled by Lu et al. 15 The proposed models were checked for their internal and external predictive power using cross validation parameters.
Experimental
Software
Geometry Optimization was performed by HyperChem (Version 7.0 Hypercube, Inc) at the AM1 level. Descriptors were calculated using HyperChem and Dragon software. 23 SPSS Software (version 13.0, SPSS, Inc.) was used for the simple multiple linear regression (MLR) analysis. PCA, ANN and PLS regression were performed in the MATLAB (Version 7.0.1 (R14), Mathworks, Inc.) environment.
Chemical data and descriptors
Compounds name and their BCFs are included in Table 1. Chemical structure of these compounds was obtained from HyperChem software and optimized on AM1 semi-empirical level. An AM1 optimization was chosen since it was developed and parameterized for common organic structures. The Optimization was preceded by the Polak-Rebiere algorithm to reach 0.01 root mean square gradient. In this study, 24 molecular descriptors including topological, 2D-autocorrelation, GETAWAY, properties and functional groups descriptors were calculated using HyperChem and Dragon software, see Abbreviations.
Non-ionic organic compounds used in this study and their experimental log BCF values.

Compounds were considered as outliers;
Compound classified in the test set.
MLR analysis
MLR analysis using the method of maximum -R 2 with stepwise selection and elimination of variables 24 was employed to model the logarithm of the BCF (log BCF) relationships with different set of descriptors to select initial input models for the artificial neural networks algorithm (ANN).
PC-ANN
Principal component analysis (PCA) and more specifically factor analysis (FA) groups together variables that are collinear to form a composite indicator capable of capturing as much of common information of those indicators as possible. Each factor reveals the set of variables having the highest association with it. The idea under this approach is to account for the highest possible variation in the indicators set using the smallest possible number of factors. Therefore, the index no longer depends upon the dimensionality of the dataset but it is rather based on the “statistical” dimensions of the data. Application of PCA on a descriptor data matrix results in a loading matrix containing factors or principal components, which are orthogonal and therefore do not correlate with each other. We used these factors as the inputs of ANN instead of the original descriptors.
In contrast to MLR, the artificial neural networks (ANN) are capable of recognizing highly nonlinear relationships. The flexibility of ANN enables it to discover more complex relationships in experimental data, when it is compared with the traditional statistical models. The PC-ANN was proposed by Gemperline 25 to improve training speed and decrease the overall calibration error.
In this method, as a preliminary treatment, the input data (i.e. molecular descriptors) was normalized so as to have zero mean and unity variance, and then were subjected to principal component analysis (PCA) before being introduced into the neural network. The most significant principal components (PCs), which explain most of the variance in the original data (>95%), were selected, ranked according to decreasing Eigen-value and then used as ANN input.
It should be noted for each model obtained with MLR separate PC-ANN models were developed so that the input's descriptors were the subsets selected by the stepwise MLR methods. In the case of each MLR model, a feed-forward neural network with back-propagation of error algorithm was constructed to model the activity structure relationships between the extracted PCs of the descriptors in one hand and the logarithm of BCF data of the non-ionic organic compounds in the other hand. More details about the model development in PC-ANN and the network architecture are explained.26–29 Over-fitting problem or poor generalization capability happens when a neural network over-learns during the training period. A too well-trained model may not perform well on unseen data set due to its lack of generalization capability. An approach to overcome this problem is the early stopping method in which the training process is concluded as soon as the overtraining signal appears. This approach requires the data set to be divided into three subsets: training, test and validation sets. The training and the validation sets are the norm in all model training processes. The test set is used to test the trend of the prediction accuracy of the model trained at some point of the training process. At later training stages, the validation error increases. This is the point when the model should cease to be trained to overcome the over-fitting problem. To achieve this purpose, the extracted PCs for each MLR model were classified into training set (60%), validation set (20%) and external test set (20%). Then, the training and validation sets were used to optimize the network performance. The regression between the network output and the activity was calculated for the three sets individually. The training function “trainscg” in MATLAB was used to train the network. To find models with lower errors, the ANN algorithm was run many times, each time run with different geometry and/or initial weights. The different ANN runs were carried out in a systematic way so that the obtained models have low training and testing root-mean-square errors (i.e. low fitness).
Partial least squares (PLS) analysis
PLS is a method for building regression models on the latent variable (LV) decomposition relating two blocks, matrices
Results and Discussion
MLR analysis
Table 2 records the regression models suggested from MLR analyses and their correlation coefficients (R). The number of descriptors in these models is varied between 3 and 15. The highest correlation coefficient obtained is 0.923 for a regression model with 15 descriptors (model
Correlation coefficient for MLR, PLS and ANN models 3-15 and cross validation parameters obtained from PLS and ANN analysis.
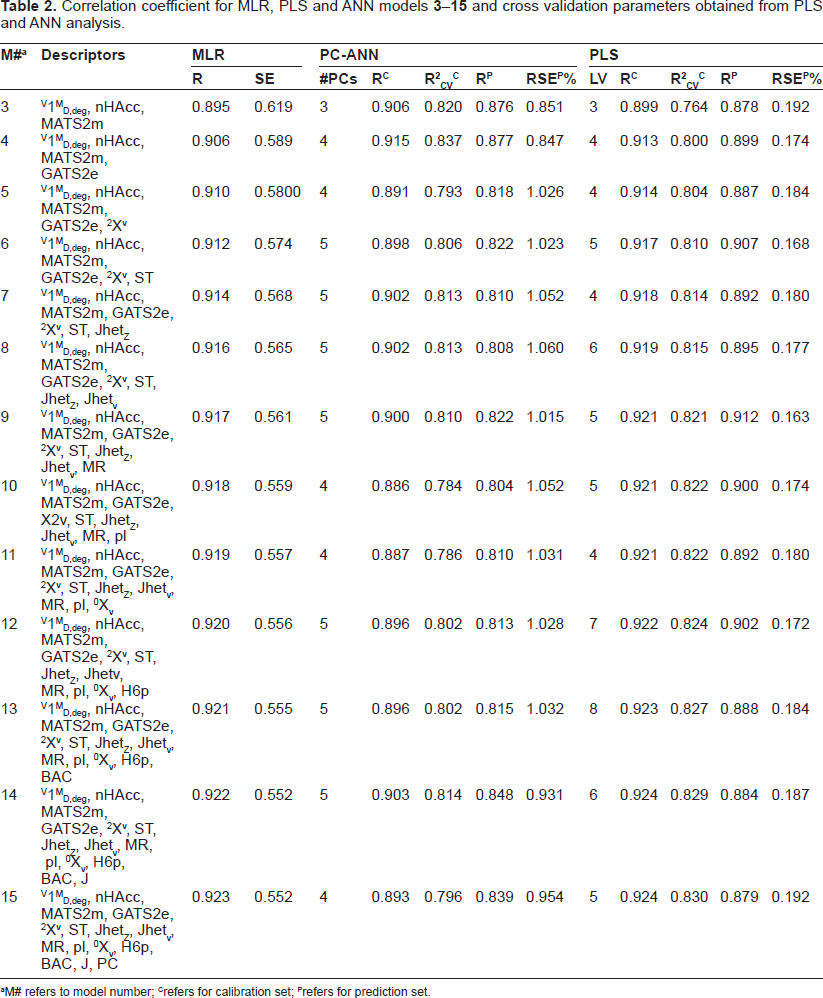
M# refers to model number;
refers for calibration set;
refers for prediction set.

Correlation of R 2 CV with MLR model number.
The regression equation for models
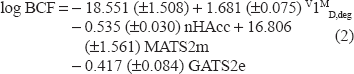
Eq. (2) shows that log BCF can be modeled by 2D autocorrelation descriptors (MATS2 m and GATS2e), the information indices descriptors (V1MD, deg) and the functional group descriptor (nHAccc). 2D autocorrekation descriptors are molecular descriptors calculated from molecular graph by summing the products of atom weights of the terminal atoms of all the paths of the considered path length (the lag). 2D autocorrelations by Moran (MATS) and Geary (GATS) algorithms are calculated from lag 1 to lag 8 for 4 different weighting schemes. The information indices descriptors are molecular descriptors calculated as information content of molecules, based on the calculation of equivalence classes from the molecular graph. Eq. (2) shows that the most significant descriptor is the Moran autocorrelation of a topological structure -lag2/weighted by atomic mass (MATS2 m) which is among the 2D autocorrelation descriptors. Furthermore, eq. (2) shows that log BCF increases with increasing MATS2M and V1MD, deg and decreases with increasing Geary autocorrelation -lag2/weighted by atomic Sanderson electronegativities (GATS2e) as well as the number of acceptor atoms for H-bonds (nHAcc) values.
Nevertheless, the number of descriptors is small according to the rule of the thumb suggested by Tute. 33 Partial least squares (PLS) and artificial neural networks algorithm (ANN) were used for further investigation of the linear and nonlinear relationships in the obtained regression models.
PC-ANN
The inputs of the ANN were the subset of the descriptors used in different MLR models (Table 2 and S1 in the supplementary material). The correlation data matrix for these descriptors is represented in supplementary material (Table S2). As it is observed, some descriptors represent high degree of collinearity. Collinear descriptors add redundancy to the input data matrix and therefore the performances of the models obtained by using these descriptors will be degraded. Therefore, we used principal components as the inputs of ANN instead of the original descriptors.
Firstly, PCA was used to classify the molecules into training, validation and prediction sets. Performing PCA overall the data set of 229 compounds and 24 descriptors and plotting the first and second principal, shows that compounds

Correlation of 1st principal component with 2nd principal component for the factor spaces of the descriptors and their BCF.
In this study, a three-layered feed-forward ANN model with back-propagation learning algorithm
34
was employed. At the first, the nonlinear relationship between the subset of descriptors selected by step-wise selection-based MLR (Table 2) and log BCF was preceded by PC-ANN models with similar structure. The number of hidden layer's nodes was set 7 for all models and the number of nodes in the input layer was the number of PCs extracted for each subset of descriptors. The results of PC-ANN modeling for MLR models number
Figure (3a) shows plots of predictive residual sum of squares (PRESS) against regression models number
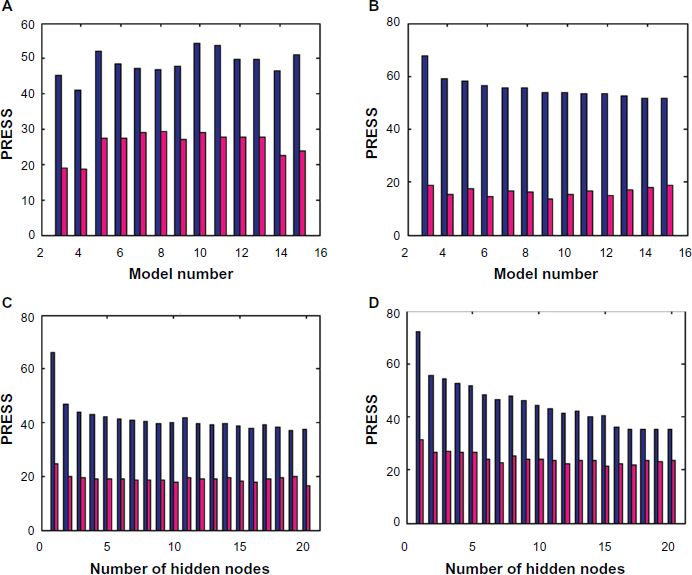
A) correlation of PRESS with ANN models (3-15). B) correlation of PRESS with PLS models (3-15). C) correlation of PRESS with different numbers of hidden nodes for ANN model 4. D) correlation of PRESS with different numbers of hidden nodes for ANN model 14.
In order to optimize the performance of the ANN models
The results for the ANN optimization for model
Figure (3d) shows plots of PRESS against number of hidden nodes for training and test sets for ANN model

Model
Table S4 in the supplementary material shows the results for randomization test that was performed to investigate the probability of chance correlation for model

Correlation of the predicted log BCF against observed one as well as their residues for A) training set. B) validation set. C) external test set of model 4 obtained by PC-ANN analysis using 10 hidden nodes.
PLS
PLS analysis with cross validation was carried out for advance investigation of the linear relationships of the obtained regression models. Model validation was achieved through leave-one-out cross-validation (LOO CV) and external validation (for a test set), and the predictive ability was statistically evaluated through the root mean square errors of calibration and validation. The calibration and prediction qualities were quantified with the correlation coefficients for the training and test sets as well as the R 2 CV (leave one out cross-validation on training set), select the LV when the R 2 CV has a high number, or determine it by computing the prediction error sum of squares (PRESS) for cross- validated models which is a standard index to measure the accuracy of a modeling method based on the cross-validation technique.
The cross-validation method employed was to eliminate only one sample at a time and then PLS calibrate the remaining standard descriptor. By using this calibration, the log BCF of the sample left out was predicted. This process was repeated until each standard had been left out once. Figure (3b) shows the associated PRESS of the training and test sets for each model. Table 2 shows that the minimum prediction error (0.163%) occurs for model 9 . The cross validation coefficient of determination for this model is high (0.821).
This model has the lowest PRESS values for the training and test sets at the same time. While other models have higher R
2
CV values than this model, they also have higher prediction errors. Accordingly, model
(Table S5) in the supplementary material shows regression and cross validation parameters for randomization test that is performed to investigate the probability of chance correlation for models

Correlation of the predicted log BCF against observed one as well as their residues for A) training set. B) external test set of model 9 obtained by PLS analysis.
This model contains the following nine descriptors: V1MD, deg, nHAcc, MATS2m, GATS2e, 2 Xv, ST, JhetZ, Jhetv, MR (see appendix) which are represented by 5 LV's.
The following conditions proposed by Golbraikh and Tropsha 37 were applied to conclude that the QSAR model has acceptable prediction power if:
where R
2
0 and R′
2
0 are the coefficients of determination characterizing linear regression with Y-intercept set at zero, the first associated with observed vs. predicted values, the second related to predicted vs. observed values; k and k’ are the slopes of the regression lines forced through zero, relating observed vs. predicted and predicted vs. observed values. Alternatively, the parameter R
2
m, where R
2
m = R2* (1-(R
2
-R
2
0))1/2, can be used.
38
This parameter penalizes a model for large differences between observed and predicted values, was also calculated. R
2
m should be larger than 0.5 for a good external prediction, which is the case for model
Comparing the linear (PLS) and nonlinear (PC-ANN) models shows that nonlinear relations improved the models over linear ones. Table 2 shows that compared with PLS and ANN results, MLR underestimate the regression coefficient values for small number of variables. Both ANN and MLR results show that Model
In summary, this study utilizes theoretical molecular descriptors to estimate BCF directly from the structure of the chemical. The most important molecular descriptors to model the BCF are topological (mainly 2d autocorrelation descriptors), geometrical (mainly encoding molecular size) and generally related to chemicals’ dimensions, polarizability, surface tension, molar ref-reactivity and number of acceptor atoms for H-bonds.
Comparison with previous studies
Table 3 summarizes the results obtained in the present study as well as other QSPR studies performed on the BCF. Zhao et al 14 performed a QSPR study on a large set of 473 compounds that was created with ISIS BASE 2.5 SP2. Wang et al 39 obtained a QSAR model by adopting topological properties and flexibility of chemicals to predict the BCF. Gramatica and Papa16,17 have performed QSAR study on the same set of non-ionic organic compounds using GA and MLR analysis without verifying the data. Fatemi et al 18 applied ANN using descriptors selected with GA but did not use PCA in the stepwise pre-selection of variables. Both Gramatica and Papa16,17 as well as Fatemi et al 18 split the data randomly, while in this contribution the data is split homogenously using the space of PCs. This implies that the training, validation and test sets include the molecules in different zones of the data distribution shown in Figure 2. Chen et al 19 developed a QSAR model for fish BCFs of 8 groups of compounds employing PLS regression, based on LSER theory and theoretical molecular structural descriptors. Their model showed that the molecular size plays a critical role in affecting the bioconcentration of organic pollutants in fish which agrees with the results found in this study where theoretical descriptors were used to develop the model and assist the mechanism interpretation. Nevertheless, the model found in this study has performed external validation in addition to the usage of a validation set of 46 compounds to detect overfitting as implemented in the early stopping method while no such procedure was used in the other studies mentioned above.
Comparison between the different QSAR models. a
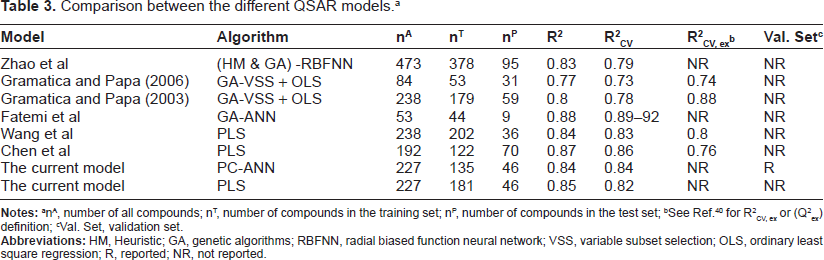
nA, number of all compounds; nT, number of compounds in the training set; nP, number of compounds in the test set;
Val. Set, validation set.
Wang et al obtained coefficients of determination of 0.80 and 0.79 for calibration and cross validation, respectively, while in this study we obtained higher values (0.85 and 0.82). Generally, the calibration and cross validation coefficient of determinations (R 2 and R 2 CV) obtained in this study (ANN model: 0.84 and 0.84/PLS model: 0.85 and 0.82) are higher than those obtained in the other studies shown in Table 3. An exception is for the R 2 and R 2 CV values obtained by Fatemi et al (0.88 and 0.89-0.92) and Chen et al (0.87 and 0.86). However, in this study, we used a larger number of compounds which implies that the results obtained in this study are more general than those obtained by Fatemi et al and Chen et al.
Conclusions
A quantitative-structural property relationship analysis has been conducted on BCF (log BCF) for 227 different non-ionic organic compounds by using PLS and the principal component-artificial neural networks modeling methods, with application of eigenvalue ranking factor selection procedure. The PLS gives improved regression models with better prediction ability compared with PC-ANN. Taking into account the complexity of the neural network based models; PLS based models are quite good to describe the QSPR of the BCF for the data set in this investigation. A 0.921 correlation coefficient was obtained using PLS with 5 LV's. The optimal model contains the descriptors V1MD, deg, nHAcc, MATS2m, GATS2e, 2 Xv, ST, JhetZ, Jhetv and MR.
Disclosure
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Acknowledgments
Omar Deeb acknowledges Prof. Dr. Jingwen Chen, Key Laboratory of Industrial Ecology and Environmental Engineering (Ministry of Education), School of Environmental Science and Technology, Dalian University of Technology (China) for his valuable suggestions to improve the manuscript. He also acknowledges Dr. Kunal Roy, Drug Theoretics and Cheminformatics Lab, Department of Pharmaceutical Technology, Jadavpur University, Kolkata (India) for his suggestions concerning validation of the proposed models.