
Abstract
The assumption of basic properties, like self-regulation, in simple transcriptional regulatory networks can be exploited to infer regulatory motifs from the growing amounts of genomic and meta-genomic data. These motifs can in principle be used to elucidate the nature and scope of transcriptional networks through comparative genomics. Here we assess the feasibility of this approach using the SOS regulatory network of Gram-positive bacteria as a test case. Using experimentally validated data, we show that the known regulatory motif can be inferred through the assumption of self-regulation. Furthermore, the inferred motif provides a more robust search pattern for comparative genomics than the experimental motifs defined in reference organisms. We take advantage of this robustness to generate a functional map of the SOS response in Gram-positive bacteria. Our results reveal definite differences in the composition of the LexA regulon between Firmicutes and Actinobacteria, and confirm that regulation of cell-division inhibition is a widespread characteristic of this network among Gram-positive bacteria.
Keywords
Introduction
The ability to uncover and decipher transcriptional regulation systems constitutes an invaluable tool in molecular biology1,2 and represents a major challenge in bioinformatics.3–5 Transcriptional regulation is mediated mainly by a subset of proteins known as transcription factors (TF) that bind DNA and can either hinder (repressors) or promote (activators) the formation of an open complex by the RNA-polymerase holoenzyme. 6 Transcription factors recognize a relatively small set of sites collectively known as the binding or sequence motif, 7 often represented using sequence logos. 8 The semi-specific recognition of binding sites by their cognate transcription factors allows implementing computational tools for the discovery and detection of transcription binding motifs and sites. 1 Motif discovery methods focus on the identification of overrepresented patterns in groups of sequences to generate a motif description.3,9 Conversely, site search algorithms take in a motif description and use pattern matching techniques to search for putative sites on DNA sequences.9,10 Scanning genomic sequences in this way leads to a noisy but informative reconstruction of transcriptional regulatory networks, which can be later validated by in vitro and in vivo methods,11–13 or linked to other sources of information in order to reconstruct regulatory networks.1,14–16
In the past, numerous studies have exploited comparative genomics approaches to analyze the composition and conservation of transcriptional regulatory networks or regulons. These studies can yield important insights into several facets of transcriptional regulatory networks that are difficult to approach experimentally. By assessing the spread of a given regulatory signal, for instance, one can infer the ancestry and biological relevance of a regulatory mechanism.17,18 Similarly, the analysis of the genetic makeup of a regulon across different species can shed light into its core evolutionary conserved components and reveal previously unidentified regulon members.11,12,19 Comparative genomics approaches to regulatory network analysis rely on the basic notion that regulons are composite entities that aggregate four different elements: the regulatory TF, a biological function, the network of regulated genes and the motif recognized by the TF. Most comparative genomics approaches to regulon analysis make the implicit assumption that both the regulatory TF and its biological function are preserved. Motif discovery techniques relying on comparative genomics typically assume also that both the network of genes and the TF-binding motif are preserved in order to apply phylogenetic footprinting techniques to enhance motif discovery.20,21 In contrast, regulon analysis techniques based on site search assume only conservation in the TF-binding motif and seek to elucidate variations in regulon composition.1,11,12,17–19,22
Forfeiting the requirement of network conservation makes site search-based analyses of regulatory networks by comparative genomics implicitly dependent on an initial description of the TF-binding motif. This description is typically based on a model organism in which a substantial number of sites12,17,19 or regulated genes17,18 is known from previous experimental work. In the latter case, a motif discovery tool is applied to gene promoter regions to generate a candidate motif to start the multi-genome search. More recently, mutational analysis of a single site has been proposed to construct a viable TF-binding motif. 13 Still, these strategies require the generation or availability of previous experimental knowledge in a model organism. This is inconvenient because this model organism can sometimes be relatively distant from the clade of interest, casting doubts on the underlying hypothesis of TF-binding motif conservation.
Many prokaryotic transcriptional networks can be described in terms of the single-input module (SIM) paradigm or as variations and elaborations of this basic configuration.23,24 In this connection paradigm, a single regulator controls the temporal activation of several cis-regulated genes. 24 It has been observed previously that the master transcription factor of a SIM is often self-regulated, 23 and that self-regulation is even more prevalent in repressor-based SIMs. 25 The bacterial SOS response is a well-known example of self-regulated SIM regulatory network. 26 The SOS response regulates a variable number of genes that are under direct transcriptional control of the LexA repressor. 27 In Escherichia coli, where the SOS response was originally described, LexA recognizes a 16 bp-long palindromic motif (CTGT-N8-ACAG). LexA dimers bind tightly to instances of this motif in the promoter region of 30 operons, regulating the activity of up to 40 genes involved in DNA repair, translesion synthesis (TLS) and regulation of cell division.22,28 In the advent of DNA damage, the recombination protein RecA acquires an active state and is able to induce self-catalytic cleavage of LexA dimers, de-repressing the SOS network.29,30 Explicit regulation of recA and self-regulation of the lexA gene ensures that repression is restored rapidly after DNA damage has been addressed. 26
The LexA protein has been shown to recognize an unusually large repertoire of binding motifs across the Bacteria domain, with more than 15 distinct motifs described to date. 27 This variety in binding motifs is associated with substantial diversity in regulon composition, which has been mapped in some bacterial classes through comparative genomics approaches.11,22 The evidence compiled thus far through experimental and in silico techniques suggests that there is a small set of genes persistently regulated by LexA in most bacteria. 27 This core LexA regulon comprises the lexA and recA genes, and is often complemented by a multiple gene cassette (imuA-imuB-dnaE2) involved in mutagenesis. 31 Recent work has analyzed the composition of the LexA regulon in two Gram-positive species (the actinobacterium Corynebacterium glutamicum 32 and the Firmicute Listeria monocytogenes), 33 complementing previous work in other Gram-positive species (Bacillus subtilis and Mycobacterium tuberculosis)34,35 and providing a multifaceted view of the Gram-positive LexA regulon.
The ever-growing abundance of genomic and meta-genomic data ensures that, within a given phylogenetic group, many sequences encoding orthologs of the same transcription factor will be readily available. By coupling the assumption of self-regulation to that of motif conservation, one can theoretically forgo the need for experimental knowledge in a model organism. Motif discovery algorithms can be applied to the upstream region of the orthologous genes encoding the transcription factor of interest in order to generate a candidate motif to conduct site search-based analysis of a simple transcriptional network. Taking advantage of the recent availability of experimental data on the SOS transcriptional network in several Gram-positive bacteria, here we provide proof of concept for this approach and we compare it against the conventional method based on extension from a single experimental model organism. Our results reveal that this approach is powerful enough to generate de novo transcriptional network maps, which can be used for functional annotation. Furthermore, we show that the use of a phylogenetically-broad sampling base for motif discovery can yield robust motifs for site search, generating more consistent results than the conventional methodology. We also show that the necessary steps of this approach can be extended to other transcriptional repressors and tool suites. Finally, we use this approach to generate for the first time a systematic mapping of the core LexA regulon in Gram-positive bacteria. This map reveals distinct patterns of LexA regulon composition between Firmicutes and Actinobacteria and supports the notion that cell-division inhibition is persistently regulated by the SOS response in these bacterial groups.
Algorithms and Datasets
Identification of Transcription Factor Homologs
Homologs for the master transcription factor of the genetic system under analysis were identified as best bidirectional BLAST hits36,37 on a balanced set of genomes from the clade of interest. This set of genomes was generated by selecting at least one, and no more than two, species for every major genus within the clade under study. The intent of this strategy was to maximize coverage while avoiding biases in representation due to the uneven distribution of sequencing projects among genera, which could distort the ensuing motif discovery process. Species were selected using the Integrated Microbial Genomes (IMG) system of the Joint Genome Institute 38 and homologues of the transcription factor were identified as best bidirectional BLAST hits using the protein sequences of well-established homologs for each phylum/class analyzed and a minimum e-value of 10–20 on the IMG BLASTP service (http://img.jgi.doe.gov/).
Motif Discovery
For each of the identified transcription factor gene homologues, the region 250 bp upstream of the predicted translation start site, which is known to harbor most promoter elements in bacteria, 39 was extracted using the IMG export service. These 250 bp regions were then fed into the MEME service of National Biomedical Computation Resource (http://meme.nbcr.net/) using any number of occurrences for a single motif, a predefined 10–20 bp motif length and otherwise standard parameters. Previous work has shown that most bacterial transcription factors target motifs in the 10–20 bp range 40 and a variable number of occurrences is required to factor in the multiplicity of binding sites described for many bacterial promoters. 41 Whenever the best motif identified by MEME was found to be a palindrome, motif discovery was repeated on a single strand with the palindrome-only option set to refine the model. Alternatively, motif discovery on these same regions was performed using the PhyloGibbs Online service (http://www.phylogibbs.unibas.ch/) using default parameters. A basic phylogeny was estimated with the WUR CLUSTALW server (http://www.bioinformatics.nl/tools/clustalw.html) using the protein sequences of the transcription factor for the CLUSTALW alignment20,42 and provided to the PhyloGibbs Online service as a Newick-formatted tree file. Because PhyloGibbs does not allow for variable motif input, we conservatively set motif width to the maximum value used for the MEME experiments (20 bp). For the purposes of the comparative genomics analysis, the model was further refined by using the MEME-inferred motif to search again the upstream regions with FITOM (see below) looking for additional binding sites. Search results with scores greater than two standard deviations below the mean of the MEME-inferred collection were considered putative binding sites and added to an expanded collection used as the standard in subsequent genomic searches.
Binding Site Search
In silico searches of putative binding sites were performed with FITOM 10 and xFITOM 43 (http://compbio.umbc.edu/software). These programs take in a collection of known sites, from which they derive a Position-Specific Frequency Matrix (PSFM). Different scoring methods based on information theory can then be applied to search a given target sequence and the programs annotate results based on the proximity of candidate sites to gene regulatory regions. The searches reported here were all conducted using the sequence information content (Ri) scoring method 44 and otherwise default parameters for FITOM/xFITOM. Searches were based on several collections of experimentally validated binding sites or on collections of binding sites inferred through motif discovery with MEME. 45 For each collection the search threshold was adjusted to eight standard deviations below the mean score for the sites present in the collection, in order to accommodate progressive threshold decrease down to six standard deviations below the mean in the comparative genomics approach (see below).
Benchmarking of the site search process was performed using collections of experimentally validated sites32–35 as reference for different genomes. For any given genome, searches were run using different collections of sites to define the search motif. Receiver Operating Characteristic (ROC) curves were then generated by plotting the percentage of experimentally validated sites (true positives) with respect to the percentage of non-experimentally validated sites (false positives) detected by the search process when using different thresholds. ROC-curves are shown only for the high-specificity thresholds typically used in site search.
Genome Sequences
A set of representative species from a phylogenetic group of interest was selected to perform the comparative genomics analyses. Representative species were chosen to include those in which binding sites for the transcription factor of interest had been experimentally reported as well as species with available reference sequences in the NCBI RefSeq database comprising all major orders within the group, while including a relatively low total number of species to allow detailed analyses of results. Genome sequences for all the selected species were downloaded from the NCBI GenBank database.
Experimental Datasets
Collections of LexA-binding sites for B. subtilis, M. tuberculosis, L. monocytogenes and C. glutamicum were compiled from experimentally validated sites reported in the literature32–35 and standardized to a length of 18 bp by searching on the reference genome and expanding, if necessary, the original site.
Comparative Genomics Analysis
To perform the comparative genomics approach, searches with the inferred binding motif were carried out using xFITOM on all genome sequences selected for analysis. Search results were then parsed sequentially, going from systematically high to low scoring sites across all genome results files. A threshold of two standard deviations below the average for the inferred binding motif was applied initially to select candidate sites. For each candidate site, homologues of the putatively regulated genes were identified as best reciprocal BLAST hits against the other selected bacterial genomes. Identified homologues were then mapped back to the corresponding results file. Whenever a gene was found to be putatively regulated in a new genome, the threshold for that particular gene was decreased by one additional standard deviation, down to a maximum of six deviations below the mean when putative evidence of regulation had been established in five or more species. This lower threshold was chosen because it identified binding sites in 95% of the gene upstream regions used for LexA motif discovery. A gene was considered to be putatively regulated if binding sites meeting the above criteria were located upstream of its orthologs in at least two different species.
46
The complete process for parsing search results files, identifying gene homologues as best reciprocal BLAST hits and assessing putative regulation (Fig. 1) was automated using custom Perl scripts. The validity of this comparative genomics approach was qualitatively assessed using the RegPredict Regulon Inference service (http://regpredict.lbl.gov/) with default parameters.
47
Schematic representation of the comparative genomics approach used in this work. (A) Motif discovery. Self-regulation is assumed for the transcription factor of interest, which is identified univocally in a particular genome. A uniform sample of genome sequences from a given phylogenetic group is selected and multiple homologues of the transcription factor are identified in these genome sequences as best-bidirectional BLAST hits using the TF of interest as the starting query. The upstream sequences of the genes coding for the TF homologues are retrieved and a motif discovery algorithm is applied to them. The resulting best motif model is refined by exploiting its palindromic nature and the collection of sites that compose it is expanded by re-searching the gene upstream regions for additional sites. (B) Comparative genomics.
Results and Discussion
Generation of the Gram-Positive LexA-binding Motif
This work explores the feasibility of applying two simple assumptions regarding a predicted transcription factor (self-regulation and motif conservation) to take advantage of the availability of genomic and meta-genomic data in order to yield a first-order map of its transcriptional network. As a test case, we use the LexA protein of Gram-positive bacteria, for which there is comprehensive experimental data available on several organisms. An obvious first step in pursuing this goal is to generate a valid candidate TF-binding motif to initiate the search procedure in target genomes. Based on the above assumptions, a candidate TF-binding motif can be obtained by applying a motif discovery algorithm to the promoter region of homologues of the transcription factor under analysis. Here we make extensive use of the standard motif discovery algorithm MEME, 45 but the same kind of analysis may be performed with motif discovery algorithms that incorporate phylogenetic information.20,21,48
To identify the LexA-binding motif, we selected a representative sample of 67 Gram-positive genomes (Supplementary data 1) and we identified LexA homologues as best-bidirectional BLAST hits using the Bacillus subtilis and Mycobacterium tuberculosis LexA proteins as queries. This resulted in the identification of 58 lexA homologues (Supplementary data 2), the upstream region of which was used for motif discovery. Default motif discovery with MEME on these 58 promoter regions yields two canonical Gram-positive LexA-binding motifs of lengths 18 and 16 bp, respectively, reported independently as the best- and second best-scoring motifs (Fig. 2A). The first motif is reported in only 38 of the 58 sequences and the second in only 32 sequences. In order to obtain a more generic motif, we expanded the best-scoring motif by conducting a conservative site search on all 58 sequences. This led to a final (expanded) collection of 71 sites distributed among 47 sequences (Fig. 2B) that was used subsequently for site search in the comparative genomics approach (Supplementary data 3). Applying a less conservative search threshold revealed that most of the lexA promoter sequences that were not represented in this expanded collection contained several putative weak sites in tandem configuration (Supplementary data 4).
LexA-binding motifs. (A) Best-scoring motif reported by MEME based on 50 sites, (B) search-expanded motif encompassing 71 sites, (C) motif from experimental sites in B. subtilis, (D) motif from experimental sites in C. glutamicum, (E) motif from experimental sites in M. tuberculosis and (F) motif from experimental sites in L. monocytogenes.
We assessed the dependency of the motif discovery approach on the number of sequences by randomly sampling the 58 promoter regions in groups of 48, 24, 12, 6 and 3 sequences and using either the MEME or PhyloGibbs motif discovery services. Even with the more restrictive parameter settings of PhyloGibbs, the LexA-binding motif was identified routinely using as few as 6 sequences, with motif discovery stabilizing fully at 48 sequences (Supplementary data 5 and Supplementary data 6). We also performed a qualitative analysis of the generality of the motif discovery approach by applying it to other self-regulated transcription factors in different phyla. We analyzed three additional transcriptional repressors (Rex, HrcA and TyrR) in, respectively, the Actinobacteria, the Firmicutes and the Gamma Proteobacteria, and we were able to infer the reported experimental motifs for all three,49–51 indicating that this type of analysis can be extended to other transcriptional systems (Supplementary data 7, Supplementary data 8, Supplementary data 9).
Search Performance of the LexA-binding Motif
Conventional approaches to regulatory network analysis by comparative genomics typically exploit experimental data, either in the form of binding site collections or known regulated genes, in a single reference species.11,12,17–19 A foreseeable problem with this approach is the progressive unreliability of the experimental motif as the phylogenetic distance between source and target species increases. Here we decided to evaluate the impact of this effect on a phylogenetically broad group of bacteria (Gram-positive bacteria) and we analyzed whether our approach, based on a single-gene multi-species derived motif, might also be subject to a similar effect.
We used published experimental results on the composition of the LexA regulatory network in four different Gram-positive bacterial species (the Firmicutes B. subtilis and L. monocytogenes, and the Actinobacteria M. tuberculosis and C. glutamicum) to benchmark the search efficiency of each of the four experimental collections, plus the MEME-derived collections, on each bacterial genome.32–35 The ROC curves shown in Figure 3 demonstrate that phylogenetic proximity does have a substantial impact on search efficiency. In all four genomes, search efficiency drops drastically when using a Firmicutes-derived motif on an Actinobacteria genome and vice versa. At the high specificities typically used for reliable site search (0.9995 specificity), sensitivity decreases by 60% on average when searching with an experimentally known motif defined in one group on a genome belonging to the other. In contrast, the automatically-derived motifs yield search efficiencies that are much closer (13% average decrease for the expanded motif) to those obtained with collections experimentally defined in the same group the searched genome belongs to (Fig. 3). The results also suggest that the expansion of the initial motif identified by MEME generates a slightly noisier motif that systematically improves search efficiency.
ROC curves for search efficiency with experimentally-validated and MEME-derived collections on four different genomes corresponding to the Firmicutes (left) and Actinobacteria (right).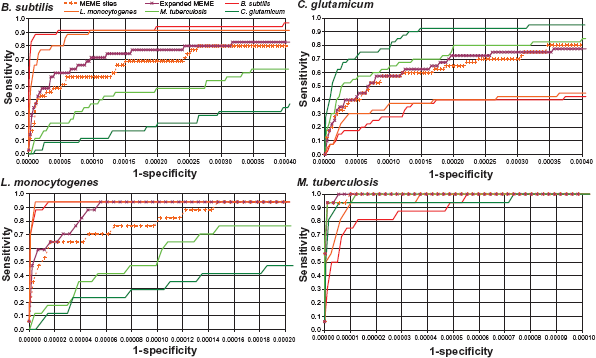
Differences in the specific shape of the Gram-positive LexA-binding motif have been noticed before,32,33,35 but their specific evolutionary relationship and their impact on search efficiency had not been assessed directly. Our results show that differences in the LexA-binding motif of Gram-positive bacteria stem mainly from the evolutionary split between Firmicutes and Actinobacteria. Furthermore, the ROC curves demonstrate that the differences observed among LexA-binding motifs have a definite impact on search efficiency. The sequence logos shown in Figure 2 illustrate how the MEME-inferred motif combines traits of both the Firmicutes (dominance of dyad central positions) and Actinobacteria (importance of spacer and adjacent positions) that allow it to perform well on both phyla. These results thus support the use of a phylogenetically broad sample for motif discovery when conducting comparative genomics analysis, as this leads to a generic motif that can achieve competitive search efficiencies in all target genomes.
Comparative Genomics of the Gram-Positive SOS Network
Composition of the LexA regulon in Gram-positive bacteria.

Only two genes, recA and lexA, are consistently detected as putatively LexA-regulated, but putative LexA-binding sites can also be detected upstream of genes coding for translesion synthesis polymerases in nearly all species. This is in agreement with the hypothesis of a conserved core SOS regulon that extends beyond recA and lexA to include TLS as a primary component of the SOS response.22,27,52–54 In this regard, it is interesting to note that SOS-induced TLS is apparently taken up by two different mechanisms in Firmicutes and Actinobacteria. In agreement with the experimental data available for individual organisms, the former appear to rely on the polymerase IV (encoded by dinB) and a polymerase V ortholog (encoded by uvrX),33,34,55 while the latter exploit error-prone nature of the second α-subunit of the DnaE polymerase (encoded by dnaE2).32,35,54 In addition, our analysis suggests that in the Actinobacteria the TLS activity of DnaE2 is quite often coordinated with expression of the mutagenic the imuA-imuB operon, which has been shown to be involved in DNA damage-inducible mutagenesis in other bacterial classes. 56 This result is consistent with the identification of SOS regulated polycistronic units encompassing imuA, imuB and dnaE2 across the Bacteria domain. 31
The comparative analyses of search efficiency reveal a consistent phylogenetic split between Firmicutes and Actinobacteria at the LexA-binding motif level (Figs. 2 and 3). This phylogenetic divide is also clearly visible in the repertoire of repair genes regulated by the SOS response in both clades (Table 1). The Firmicutes, for instance, maintain LexA-regulation of the excision repair uvrBA operon, a canonical element of the E. coli SOS regulon. In contrast, LexA-regulation of uvrB and uvrA is absent in the Actinobacteria, where the uvrBA operon organization has been disrupted. The lack of LexA regulation for the excision repair system in Mycobacteria has been noticed before, even though these and other repair genes area induced by DNA damage. 57 Our findings indicate that lack of LexA regulation for uvrA and uvrB is the norm among Actinobacteria, leaving open the question of how, if at all, these genes are regulated by DNA damage in this clade. 58
In contrast with the Actinobacteria, the Firmicutes appear to lack regulation of another hallmark of the E. coli SOS regulon: the Holliday junction complex encoded by the ruvAB operon. In this case, the absence of LexA regulation is associated with the absence of the ruvC gene, which heads the ruvCAB operon in Actinobacteria. Regarding the clear-cut phylogenetic split between both groups when analyzing regulon organization, it is also worth noting that genes regulated by LexA only in the Actinobacteria (eg, splB, alk, ruvC, whiB2) are usually absent in the Firmicutes. In contrast, genes under LexA regulation only in the Firmicutes (yhaO, uvrB, pcrA) are typically present, but not regulated by LexA, in the Actinobacteria. This is consistent with a RecA-independent mechanism of DNA damage-induction in the Actinobacteria, which has already been shown to coordinate the expression of several DNA repair genes in the Mycobacteriaceae. 57
Regulation of Cell Division by the SOS Response
In E. coli, the SOS response regulates cell division by blocking the formation of the FtsZ ring via the product of the sulA gene. 59 Later research has shown that in many bacterial species the sulA gene is frequently found in an operon with lexA,31,46 providing a straightforward means for its regulation by the SOS response. In 2003, the protein encoded by the B. subtilis yneA gene, which forms a divergent gene pair with lexA, was shown to inhibit cell division upon induction of the SOS response. 60 This finding was remarkable because YneA is structurally different and phylogenetically unrelated to SulA. More recently, the products of two additional genes forming a divergent gene pair with lexA (Rv2719c in M. tuberculosis and divS in C. glutamicum) have also been shown to suppress cell division upon induction of the SOS response.61,62 Both the B. subtilis YneA and the M. tuberculosis Rv2719c contain a peptidoglycan-binding LysM domain that has been shown to be necessary for suppression of cell division by YneA, but not by Rv2719c.62,63 In contrast, the C. glutamicum DivS does not contain a peptidoglycan-binding LysM domain, indicating that these three proteins interfere with cell division in different ways.
Our analysis suggests that this trend towards regulation of cell division by the SOS response through divergent pairing of a cell division suppressor with lexA is most likely a defining trait of Gram-positive bacteria. Beyond the above cases and the L. monocytogenes yneA ortholog,
33
the comparative genomics analysis identifies two additional genes containing a LysM domain (Acel_1478 in A. cellulolyticus and Lxx15870 in L. xyli) divergently paired with lexA. These two genes do not present significant sequence similarity with either yneA or Rv2719c. However, the presence of a conserved LysM domain and the conservation of synteny (Fig. 4) suggest that their function is likely to be preserved. Synteny is also maintained in the N. farcinica NFA_37990 and S. aureus SAV1340 genes, but neither presents a conserved domain. Nonetheless, given that three independent mechanisms for cell-division suppression have already been suggested for genes paired divergently with lexA, the potential role of these two genes in cell division should be an interesting target for experimental analysis.
Schematic representation of the genomic region encompassing the lexA gene and divergently paired putative and known (yneA, divS, Rv2719c) cell division inhibitors.
Conclusion
This work analyzes the feasibility of exploiting basic assumptions of simple transcriptional networks in order to infer regulatory motifs from the vast amount of genomic and meta-genomic data available, and to reconstruct regulatory networks through comparative genomics using the inferred motif. Our results provide proof of concept for this approach using the SOS regulatory network of Gram-positive bacteria as a test case, paving the way for the development of automated methods that make use of the overabundance of sequence data for de novo inference of simple regulatory networks. Furthermore, benchmarking against experimental data suggests that inferred motifs may yield more robust search patterns. The analysis of the SOS response in Gram-positive bacteria shows clear differences in the composition of the LexA regulon between the two main groups of Gram-positive bacteria and reinforces the notion that part of the DNA repair machinery of the Actinobacteria is regulated independently of LexA. Finally, our study suggests that regulation of cell-division by the SOS response is prevalent in Gram-positive bacteria, providing further evidence of convergent evolution of this trait and pointing to interesting candidates for experimental research.
Author Contributions
Conceived and designed the experiments: IE. Analysed the data: JPC, FM, JRT, IE. Wrote the first draft of the manuscript: IE. Contributed to the writing of the manuscript: JPC, IE. Agree with manuscript results and conclusions: JPC, FM, JRT, IE. Jointly developed the structure and arguments for the paper: JPC, JRT, IE. Made critical revisions and approved final version: JPC, IE. All authors reviewed and approved of the final manuscript.
Funding
This work was supported by the UMBC Office of Research through an Special Research Assistantship/Initiative Support (SRAIS) award.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Footnotes
Supplementary Data
Representative genomes from Gram-positive species selected to identify lexA homologs. The IMG taxon ID, species name and genome status are provided for each genome.
Homologs of lexA identified through best-reciprocal BLAST hit in the selected species. The table displays basic gene and protein information, as well as the –250 bp upstream sequence for each gene.
Text file containing the collection of 71 LexA-binding sites generated by expansion of the initial MEME motif using site search on the upstream region of indentified lexA homologs.
Table of putative LexA-binding sites located in the promoter region of the 58 lexA upstream regions. Multiple instances of weak (from 3 down to 6 standard deviations below the mean; 11.37–5.45 bits) sites for a single promoter are highlighted.
Tables summarizing five independent MEME motif discovery runs on randomly sampled subsets of the collection of 58 lexA homologues upstream sequences (Supplementary data 2), for decreasing subset sizes of 48, 24, 12, 6 and 3 sequences.
Tables summarizing five independent PhyloGibbs motif discovery runs on randomly sampled subsets of the collection of 58 lexA homologues upstream sequences (Supplementary data 2), for decreasing subset sizes of 48, 24, 12, 6 and 3 sequences.
Results for MEME motif discovery on homologues of the TyrR repressor in the Gamma-Proteobacteria. The selected list of species and upstream regions for motif discovery are provided together with the three best results of the motif discovery process.
Results for MEME motif discovery on homologues of the Rex repressor in the Actinobacteria. The selected list of species and upstream regions for motif discovery are provided together with the three best results of the motif discovery process.
Results for MEME motif discovery on homologues of the HrcA repressor in the Firmicutes. The selected list of species and upstream regions for motif discovery are provided together with the three best results of the motif discovery process.
ROC curves for search efficiency with experimentally-validated and MEME-derived collections on four different genomes corresponding to the Firmicutes (left) and Actinobacteria (right). Sensitivity corresponds to the fraction of experimentally validated binding sites detected by the search algorithm. Specificity is the fraction of the rest of genomic positions reported by the search algorithm.
Table of indentified LexA-binding sites. The regulated gene name, product and GenBank locus number, as well the score, sequence, strand and distance to the gene translation start site are provided for all identified LexA-binding sites.
Results table for the comparative genomics analysis of the LexA regulon in Gram-positive bacteria with the RegPredict service, using the expanded MEME collection as the search motif and otherwise default parameters. Accession numbers indicate the presence of homologs in each species. Predicted LexA regulation is indicated colored boxes, which denote, respectively, the presence of a strong, weak or multiple LexA site, or regulation inferred from predicted membership in a regulated operon. Empty cells indicate that a given gene is absent from a particular genome. Species abbreviations are as follows: Bsu–-B. subtilis, Cac–-C. acetobutylicum, Efa–-E. faecalis, Lmo–-L. monocytogenes, Sau–-S. aureus, Ace–-A. cellulolyticus, Cgl–-C. glutamicum, Lxy–-L. xyli, Mtu–-M. tuberculosis, Nfa–-N. farcinica and Sgr–-S. griseus. Subsequent pages provide details concerning the regulated genes and the identified sites.
Acknowledgements
Writing of this paper was supported by a UMBC SFF award. FM was supported by the UMBC Meyerhoff Scholars Program.
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.